
Las presentaciones clínicas varían mucho, desde leves hasta potencialmente mortales.
La histiocitosis, también conocida como histiocitosis de células de Langerhans (HCL), y formalmente llamada histiocitosis X, representa un grupo de trastornos poco frecuentes que involucran células específicas que normalmente tienen un papel importante como parte del sistema inmune.
Las histiocitosis abarcan un grupo de diversos trastornos caracterizados por la acumulación e infiltración de números variables de monocitos, macrófagos y células dendríticas en los tejidos afectados.
Tal descripción excluye enfermedades en las que la infiltración de estas células se produce en respuesta a una patología primaria.
Aunque ha pasado casi un siglo desde que se reconocieron los trastornos histiocíticos, su fisiopatología ha comenzado a dilucidarse con la aplicación de análisis moleculares.
En los últimos 50 años, la nomenclatura utilizada para describir los trastornos histiocíticos ha cambiado sustancialmente para reflejar la amplia gama de manifestaciones clínicas y la gravedad clínica variable de algunos trastornos que tienen los mismos hallazgos patológicos.
Por ejemplo, la entidad ahora denominada histiocitosis de células de Langerhans se dividió inicialmente en granuloma eosinofílico, enfermedad de Hand-Schüller-Christian y enfermedad de Abt-Letterer-Siwe, dependiendo de los sitios y la gravedad.
Más tarde, se descubrió que eran manifestaciones de una sola entidad y se unificaron bajo el término histiocitosis X.
Más recientemente, esta designación se cambió a histiocitosis de células de Langerhans en base a la sugerencia de Nezelof de que la célula de Langerhans representaba la célula primaria implicada en la fisiopatología de la enfermedad.
Fisiopatología
La mejor comprensión de la patología de los trastornos histiocíticos requiere el conocimiento de los orígenes, la biología y la fisiología de las células involucradas.
Los histiocitos normales se originan a partir de células madre pluripotentes, que se pueden encontrar en la médula ósea. Bajo la influencia de diversas citocinas, como por ejemplo:
Factor estimulante de colonias de granulocitos y macrófagos (GM-CSF, por sus siglas en inglés). Factor de necrosis tumoral alfa (TNF-alfa, por sus siglas en inglés). Interleucina [IL] -3, IL-4.
Estas células precursoras pueden convertirse en comprometido y diferenciado para convertirse en un grupo específico de células especializadas.
Las células madre comprometidas pueden madurar para convertirse en células de procesamiento de antígenos, y algunas poseen capacidades fagocíticas.
Estas células incluyen macrófagos tisulares, monocitos, células dendríticas, células reticulares interdigitantes y células de Langerhans.
Las células madre pluripotentes también pueden comprometerse a producir células dendríticas. Cada categoría de histiocitosis se puede rastrear a la proliferación reactiva o neoplásica en uno de estos linajes celulares.
La importancia de las células dendríticas para presentar antígenos a los linfocitos T y B se reconoce cada vez más. Las células dendríticas parecen desarrollarse en varias vías.
Las células dendríticas inmaduras responden al factor estimulante de colonias de granulocitos-macrófagos (no al factor estimulante de colonias de macrófagos [M-CSF]) y se comprometen a generar células dendríticas, que son células presentadoras de antígenos «profesionales» (CPA, por sus siglas en inglés).
Estas células pueden capturar el antígeno y migrar a los órganos linfoides, donde presentan los antígenos a las células T vírgenes. Las células dendríticas también son estimuladores eficientes de los linfocitos de células B.
La inducción efectiva de respuestas de células T específicas de antígeno requiere interacción entre las células dendríticas y los linfocitos T para cebar a las últimas células para su expansión y respuestas inmunes posteriores.
La superficie de la célula presentadora de antígeno contiene 2 proteínas de unión a péptido: es decir, complejo principal de histocompatibilidad (MHC, por sus siglas en inglés) clases I y II.
Que pueden estimular: células T citotóxicas (TC), células reguladoras T (Treg, por sus siglas en inglés) y células auxiliares T (TH, por sus siglas en inglés).
Aunque los linfocitos circulantes de células T pueden reconocer antígenos de forma independiente, su número es pequeño.
Las células dendríticas muestran una gran cantidad de complejo peptídico complejos de histocompatibilidad en su superficie y pueden aumentar la expresión de receptores coestimuladores y migrar a los ganglios linfáticos, el bazo y otros tejidos linfoides, donde activan células T específicas.
La primera señal puede implicar la interacción entre un péptido complejo de histocompatibilidad mayor complejo I y/o complejo de histocompatibilidad principal II en una célula presentadora de antígeno con el receptor de células T (RCT) en los linfocitos efectores.
Los receptores de células T pueden reconocer fragmentos de antígeno unidos al complejo principal de histocompatibilidad en la superficie de una célula presentadora de antígeno.
La interacción coestimuladora (es decir, la segunda señal) está entre CD80 (B7.1)/CD86 (B7.2) en la célula dendrítica, y CD28 en las células T.
Una combinación de las 2 señales activa la célula T, dando como resultado una regulación positiva de la expresión de CD40L, que, a su vez, puede interactuar con el receptor CD40 expresado en células dendríticas.
En ratones deficientes en perforina, la producción de citocinas anormalmente potenciada por las células T se debe a la sobreestimulación de las células presentadoras de antígeno después de una infección viral.
Esta interacción de célula a célula entre células dendríticas y células T genera una respuesta de células T específica de antígeno.
Se presume que la función efectiva de la presentación del antígeno por las células dendríticas refleja que estas células, además de las moléculas complejas de histocompatibilidad principales, expresan una alta densidad de otros factores coestimuladores.
Las células dendríticas pueden producir varias citocinas, incluida IL-12, que es fundamental para el desarrollo de células TH 1 de células T CD4 + vírgenes.
La ligación de CD40 en las células dendríticas desencadena la producción de grandes cantidades de IL-12, lo que mejora la capacidad estimulante de las células T.
Esta observación sugiere que la retroalimentación a las células dendríticas da como resultado señales que son críticas para la inducción de respuestas inmunes.
La naturaleza de la última interacción y el requerimiento de una activación óptima de células dendríticas no se comprende completamente.
Las células dendríticas en cultivo derivadas de monocitos de sangre humana expuestos a factor estimulante de colonias de granulocitos-macrófagos e IL-4 seguido de maduración en un medio acondicionado con monocitos han aumentado la actividad presentadora de antígeno.
Los medios acondicionados con monocitos contienen factores críticos de maduración que contribuyen a este proceso.
Las células dendríticas están presentes en los tejidos en estado de reposo y no pueden estimular las células T. Su función es capturar y fagocitar antígenos que, a su vez, inducen su maduración y movilización.
Las células dendríticas inmaduras residen en la sangre, los pulmones, el bazo, el corazón, los riñones y las amígdalas, entre otros tejidos. Su función es capturar el antígeno y migrar a los órganos linfoides que drenan para cebar las células T CD4+ y CD8+.
En el proceso de su función, estas células maduran y aumentan su capacidad de expresar receptores coestimuladores y disminuir su capacidad para procesar antígenos.
Estas células pueden fagocitar, formando vesículas pinocíticas para muestrear y concentrar su medio circundante, que se llama macropinocitosis.
Las células dendríticas inmaduras expresan receptores que median la endocitosis, incluidos los receptores de lectina de tipo C, como el receptor de manosa macrófago y los receptores DEC205, FC-gamma y FC-épsilon.
Los componentes microbianos, así como IL-1, factor estimulante de colonias de granulocitos y macrófagos y factor de necrosis tumoral alfa, tienen un papel importante en la respuesta celular y pueden estimular la maduración de las células dendríticas, mientras que la IL-10 se opone.
Las células dendríticas maduras poseen numerosos procesos finos (velos, dendritas) y tienen una movilidad considerable.
Estas células, ricas en las principales clases de complejos de histocompatibilidad I y II, tienen abundantes moléculas para la unión y coestimulación de las células T, que implica CD40, CD54, CD58, CD80/B7-1 y CD86/B7-1.
Las células dendríticas maduras expresan altos niveles de IL-12. Altos niveles de CD83 (un miembro de la superfamilia de inmunoglobulinas [Ig]) y p55 o fascin (una proteína que agrupa actina) están presentes en estas células, a diferencia de los bajos niveles que están presentes en las células inmaduras.
IL-1 mejora la función de la célula dendrítica. Este efecto parece ser indirecto y debido a la activación de factores asociados al receptor del factor de necrosis tumoral (TRAF).
Las células dendríticas maduras también expresan altos niveles de la familia de proteínas de control transcripcional NF-kappaB.
Estas proteínas regulan la expresión de varios genes que codifican proteínas inflamatorias e inmunes.
La señalización por medio de la familia del receptor del factor de necrosis tumoral (por ejemplo, factor de necrosis tumoral-R, CD40, citocina inducida por activación del factor de necrosis tumoral [TRANCE], activador del receptor de NF-kappaB [RANK]) activa NF-kappaB.
La respuesta inmunológica de las células dendríticas a un antígeno dado implica en parte el desencadenamiento de vías de transducción de señales que involucran a la familia del factor de necrosis tumoral R y factores asociados al receptor del factor de necrosis tumoral.
La información sobre el destino de las células dendríticas después de estos eventos es escasa. Las células dendríticas desaparecen de los ganglios linfáticos 1-2 días después de la presentación del antígeno, posiblemente debido a la apoptosis.
La proteína CD95 (Fas) se sugiere que tenga un papel en la muerte de la célula dendrítica. Sin embargo, aunque las células dendríticas expresan CD95, la ligación de CD95 no induce la apoptosis.
Los experimentos indican que las células dendríticas inmaduras son parcialmente susceptibles a la apoptosis mediada por el receptor de la muerte. El ligando inductor de apoptosis relacionado con el factor de necrosis tumoral (TRAIL) se puede unir a 5 receptores separados.
Los dominios funcionales de muerte citoplásmica caracterizan los receptores TRAIL-R1, los receptores TRAIL-R2 y los receptores CD95. Por el contrario, TRAIL-R3 es un receptor truncado anclado a la membrana, y TRAIL-R4 no tiene un dominio de muerte funcional.
Las células dendríticas expresan CD95, TRAIL-R2 y TRAIL-R3 en niveles comparativos. Similar al papel de CD95L, el de la apoptosis mediada por TRAIL de células dendríticas maduras ha sido controvertido.
También se han publicado datos sobre la apoptosis mediada por TRAIL en estas células, aunque estos datos siguen siendo controvertidos. Las células dendríticas maduras suelen ser resistentes a la apoptosis mediada por TRAIL y CD95L.
C-FLIP, que es la proteína inhibidora de caspasa-8 capaz de inhibir la apoptosis mediada por el receptor de muerte, se expresa altamente en células dendríticas maduras, mientras que solo se encuentran niveles bajos en células inmaduras.
La sobreexpresión de C-FLIP inhibe las señales del receptor de la muerte. La expresión de C-FLIP en células dendríticas se regula positivamente durante la maduración.
Tenga en cuenta que la participación de CD95 en células dendríticas inmaduras por CD95L induce la maduración fenotípica y funcional de estas células.
Además, una célula dendrítica activada por CD95 regula por incremento la expresión del complejo mayor de histocompatibilidad clase II y receptores coestimuladores, que es esencial para la función de estas células.
Además, tal interacción regula positivamente la expresión de la proteína de membrana asociada a lisosomas de células dendríticas (DC-LAMP) y provoca la secreción de citoquinas proinflamatorias, que incluyen IL-1 beta y TNF-alfa.
Algunos artículos sugieren la clasificación de la histiocitosis de células de Langerhans de alto riesgo (HCL, por sus siglas en inglés) como una neoplasia mieloide y la hipótesis de que la enfermedad de alto riesgo surge de la mutación somática de un progenitor hematopoyético.
Algunos autores proponen que la enfermedad de bajo riesgo surge de la mutación somática de las células dendríticas precursoras restringidas a los tejidos.
Estas hipótesis se basan en el hallazgo de la mutación BRAF-V600E en las fracciones circulantes CD11C (+) y CD14 (+) y en las células progenitoras hemopoyéticas CD34 (+) de la médula ósea.
Por otro lado, la mutación se limitó a células dendríticas lesionales CD207 (+) en pacientes con histiocitosis de células de Langerhans de bajo riesgo.
Actualmente, se cree que la histiocitosis de células de Langerhans surge de la proliferación de células de Langerhans y dendric, que normalmente están restringidas a la piel y linfáticos.
La función de las células normales de Langerhans es la inmunosupervisión cutánea. Estas células pueden migrar a los ganglios linfáticos regionales y presentar antígeno potencialmente a las células T paracorticales y provocar su transformación en células dendríticas interdigitantes.
Algunas células cancerígenas interrumpen la función de las células dendríticas, bloquean el desarrollo de respuestas inmunes específicas del tumor y permiten que los tumores eviten el reconocimiento.
Para contrarrestar este efecto, las células dendríticas pueden producir la proteína antiapoptótica Bcl-xL.
La estimulación de las células dendríticas por CD154, IL-12 o IL-15 aumenta la expresión de Bcl-xL. La información obtenida de la fisiología normal de las células dendríticas puede conducir potencialmente a modalidades de tratamiento para los trastornos histiocíticos.
Síntomas de la histiocitosis
Si bien se desconoce la causa de la histiocitosis de células de Langerhans, la histiocitosis de células de Langerhans con frecuencia puede comportarse como cáncer y, por lo tanto, es tratada por especialistas en cáncer.
Un histiocito es una célula inmunitaria normal que se encuentra en muchas partes del cuerpo, especialmente en la médula ósea, el torrente sanguíneo, la piel, el hígado, los pulmones, las glándulas linfáticas y el bazo.
En la histiocitosis, los histiocitos se mueven a los tejidos donde normalmente no se encuentran y causan daño a esos tejidos. Algunas formas son genéticas.
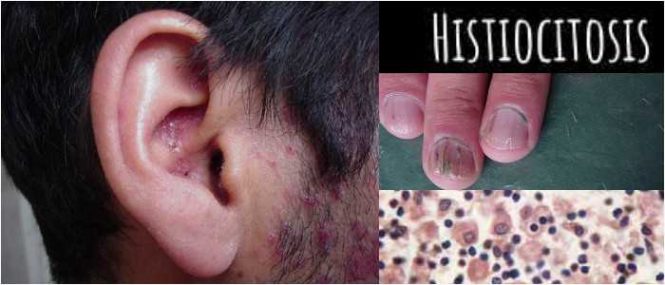
El primer signo de histiocitosis suele ser una erupción en el cuero cabelludo. Puede haber dolor en un hueso, secreción del oído, pérdida de apetito y fiebre. A veces el estómago está hinchado y doloroso.
Ocasionalmente, un área del cerebro conocida como glándula pituitaria se ve afectada, y esto puede provocar que el niño pase grandes cantidades de orina y tenga mucha sed.
Otros posibles signos y síntomas incluyen: pérdida de peso, ictericia, vómitos, cojera, baja estatura, retraso en la pubertad, deterioro mental, dolor de cabeza, mareos, convulsiones, protuberancias en los ojos y/o erupción generalizada.
Piel: los bultos rojos y escamosos en los pliegues de la piel son comunes. Los bebés con histiocitosis de células de Langerhans pueden tener cueros cabelludos rojos y escamosos, lo que a menudo se confunde con la caspa, una afección cutánea común.
Hígado: usualmente, solo los casos severos de histiocitosis de células de Langerhans afectan el hígado. Su piel puede aparecer con ictericia o amarillo, y su sangre puede tardar más en coagularse.
Ganglios linfáticos: estas glándulas, que están detrás de las orejas, en el cuello y en otros lugares, pueden hincharse. También es posible que tenga problemas para respirar o tenga tos.
En los niños, la histiocitosis generalmente afecta a los huesos y puede consistir en sitios únicos o múltiples. El cráneo se ve frecuentemente afectado.
Los tumores producen una apariencia «punzonada» en los rayos X de los huesos. A veces, los niños experimentan fracturas espontáneas como resultado de estas lesiones óseas.
Los niños mayores de cinco años generalmente tienen la enfermedad del sistema único, con solo afectación ósea. Los niños pequeños, especialmente los bebés, son más propensos a tener la enfermedad multisistémica.
La mayoría de los casos de histiocitosis afectan a niños entre las edades de uno y 15 años, aunque personas de todas las edades pueden desarrollar histiocitosis de células de Langerhans.
La incidencia alcanza un pico entre los niños de entre 5 y 10 años. Se cree que la histiocitosis afecta aproximadamente a una a dos de cada 200,000 personas cada año. La causa exacta de la histiocitosis es desconocida.
Sin embargo, estudios recientes indican que es causada por el desarrollo y la expansión de una célula de Langerhans anormal que posteriormente conduce a la acumulación de otras células del sistema inmune, lo que produce colecciones o tumores en diversas áreas del cuerpo.
Causas de la histiocitosis
No conocemos todas las razones por las que algunas personas padecen histiocitosis de células de Langerhans. Alrededor de la mitad de las personas con el trastorno tienen un gen defectuoso que hace que las células inmunes de Langerhans crezcan fuera de control.
Esa mutación genética ocurre después del nacimiento, lo que significa que generalmente no obtendrás histiocitosis de células de Langerhans de tus padres. Los investigadores sospechan que otras cosas también pueden jugar un papel:
- Fumar, padres que estuvieron expuestos a toxinas ambientales, como benceno o polvo de madera, infecciones como un recién nacido y antecedentes familiares de enfermedad tiroidea.
Pruebas de diagnóstico de la histiocitosis
La histiocitosis de células de Langerhans a menudo se clasifica como un solo sistema, cuando la enfermedad afecta solo una parte del cuerpo, o multisistema, cuando afecta a más de una parte del cuerpo.
Las pruebas de diagnóstico para niños pueden incluir:
Una biopsia, en la cual se toma una pequeña muestra de piel y/o hueso y se examina bajo un microscopio para detectar células anormales (Biopsia de la piel y de médula ósea para verificar si hay células de Langerhans).
Radiografías de todos los huesos del cuerpo para descubrir cuántos huesos están afectados y escaneos de rutina de los huesos, el cráneo y los pulmones; y análisis de sangre (hemograma completo) y prueba de una mutación genética en BRAF V600E.
Estas pruebas ayudarán al médico a determinar si la enfermedad es del tipo de sistema único o multisistémico.
Se puede hacer una radiografía de todo el sistema esquelético para determinar qué tan extensa es la enfermedad y si está indicada o no la afección sistémica.
Las pruebas de diagnóstico para adultos también pueden incluir:
- Broncoscopia con biopsia.
- Radiografía de pecho.
- Pruebas de función pulmonar.
La histiocitosis de células de Langerhans a veces está relacionada con el cáncer. Se deben realizar tomografías computarizadas y biopsia para descartar un posible cáncer.
Tratamiento de la histiocitosis
Dependiendo de la extensión de la enfermedad, la histiocitosis de células de Langerhans a menudo se trata con quimioterapia y esteroides para suprimir la función del sistema inmune y la producción de histiocitos.
La duración del tratamiento variará de niño a niño. Muchos pacientes son elegibles para ensayos institucionales internacionales y locales.
La radioterapia, el tratamiento con rayos X específicos o la cirugía limitada también se pueden usar para tratar lesiones óseas en algunas situaciones.
La mayoría de los pacientes que desarrollan histiocitosis tienen recuperaciones completas. A veces, la enfermedad puede reaparecer, por lo que el paciente tendrá visitas de seguimiento regularmente programadas en la clínica para pacientes ambulatorios como precaución.
Investigación de la histiocitosis
Se están probando nuevas ideas para determinar las causas de la histiocitosis de células de Langerhans y también por qué algunos pacientes responden mejor que otros al tratamiento.
Se están desarrollando nuevos tipos de terapias, que incluyen nuevos tipos de fármacos, así como enfoques que dirigen anticuerpos o moléculas pequeñas a la célula de Langerhans anormal sin afectar a los tejidos normales.
Epidemiología
La incidencia de histiocitosis de células de Langerhans es de 4-10 por millón de habitantes. Sin embargo, debido a que muchas lesiones óseas y cutáneas pueden no ser diagnosticadas como histiocitosis de células de Langerhans, esta tasa puede ser una subestimación.
La incidencia estimada de histiocitosis de células de Langerhans neonatal, determinada mediante el Registro de Cáncer de la Infancia Alemana basado en la población, es de 1 a 2 por millón de recién nacidos.
Morbilidad y mortalidad
Sexo
La proporción total de hombres a mujeres es de 1.5: 1. La proporción hombre-mujer en individuos que tienen afectación del sistema de un solo órgano es de 1.3: 1, y la relación hombre-mujer en individuos con enfermedad multisistémica 1.9: 1.
Años
La histiocitosis de células de Langerhans puede ocurrir en individuos de cualquier edad. La incidencia alcanza su punto máximo en niños de 1 a 3 años. En un estudio, la edad al momento del diagnóstico fue de 0.09-15.1 años.
Los pacientes con afectación única del sistema fueron mayores que aquellos con afectación multisistémica. Los casos fetales y neonatales, aunque son raros, pueden ocurrir.