
Hablamos de una proteína citoesqueletica bastoniforme de gran tamaño.
En 1868, Duchenne de Boulogne describió clínicamente una enfermedad progresiva responsable de una distrofia muscular que hoy lleva su nombre, sin comprender su origen.
Sobre esta distrofia muscular hizo posible identificar el gen y la proteína resultante fue llamada entonces: distrofina.
Estructura de la distrofina
La distrofina es una proteína que está asociada con una compleja transmembrana de células del músculo esquelético y constituye el 0.01% de la proteína muscular total y el 5% de las proteínas del citoesqueleto sarcolémico.
Se encontró que la distrofina está asociada con la membrana plasmática tanto por estudios de fraccionamiento subcelular como por inmunotinción de criosecciones de biopsia muscular.
Los estudios de microscopía de inmunoelectrón mostraron que la distrofina estaba en la cara intracelular de la membrana plasmática, con cierta periodicidad que sugiere una red.
La distrofina se relaciona con la membrana a través de una serie de proteínas asociadas a ella.
La distrofina se une a la actina filamentosa en múltiples sitios a lo largo de la molécula, con los sitios de interacción más fuertes en el dominio de unión a actina amino terminal de la proteína.
Al vincular la red de actina intracelular con la membrana plasmática y la lámina basal extracelular, el citoesqueleto de membrana basado en distrofina probablemente protege a la membrana del daño inducido por la contracción.
Tanto los sarcoglicanos como los distroglicanos muestran reducciones secundarias en los niveles de proteína en pacientes con distrofinopatía.
Las mutaciones en cualquiera de los cuatro genes sarcoglicanos causan distrofias autosómicas recesivas que son clínicamente similares a la distrofia muscular de Duchenne.
Función
El complejo de distrofina estabiliza la membrana plasmática de las células musculares estriadas.
Las mutaciones de pérdida de función en los genes que codifican la distrofina, o las proteínas asociadas, desencadenan la inestabilidad de la membrana plasmática y la pérdida de miofibras.
Las mutaciones en la distrofina se han catalogado ampliamente, proporcionando una notable correlación estructura-función entre la estructura proteica predicha y los resultados clínicos.
La restauración de la distrofina es probablemente más compleja, debido al papel del complejo de distrofina como un amplio integrador del citoesqueleto.
Ubicación de la distrofina
La distrofina se localiza en el aspecto interno del sarcolema y es abundante en la unión miotendinosa y en la membrana postsináptica de la unión neuromuscular.
La distrofina forma parte integral del citoesqueleto de un músculo y conecta el aparato contráctil con el sarcolema.
La distrofina se expresa principalmente en las células musculares esqueléticas, cardíacas y lisas, con cantidades más pequeñas expresadas en el cerebro y la retina.
Las isoformas de la distrofina , que son más pequeñas en tamaño, se expresan en casi todos los tejidos examinados.
La distrofina en el cerebro es importante en el mantenimiento de la sinapsis; la deficiencia de la isoforma cerebral de la distrofina se asocia con deficiencias cognitivas observadas en pacientes con mutaciones de distrofina.
Las deleciones o anomalías del gen de la distrofina causan una ausencia o deficiencia de distrofina , lo que da como resultado las distrofias musculares de Duchenne y Becker ligadas al cromosoma X.
Consecuencias de una alteración de la distrofina
Ya sea una ausencia total o una deficiencia parcial (proteína truncada y / o en cantidad disminuida) en la distrofina, las consecuencias se revelan de varias maneras.
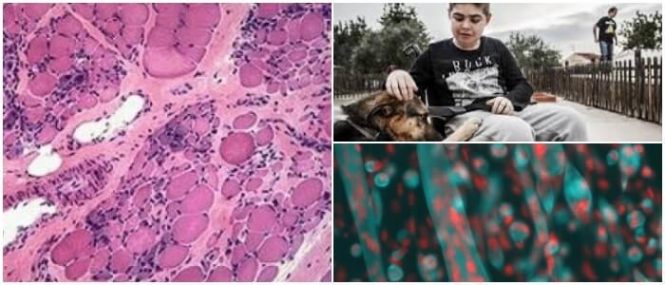
Para el músculo esquelético se pierde la integridad de la membrana de las fibras musculares responsables de la distrofia muscular.
En el músculo cardíaco, la expresión anormal de la distrofina es responsable de la cardiomiopatía aislada o, en la mayoría de los casos, asociada con la distrofia muscular.
Además, cabe señalar que existen cardiomiopatías relacionadas con la ausencia de distrofina tanto en pacientes con distrofia muscular de Duchenne.
En menor medida, la deficiencia de distrofina en el músculo liso puede llevar a consecuencias vasculares y digestivas.
La distrofina también está presente en 5 isoformas diferentes en el sistema nervioso central y 2 isoformas en la retina.
Las mutaciones en el gen de la distrofina son responsables de la mayoría de los pacientes con trastornos cognitivos y / o neuropsiquiátricos más o menos graves, según el número de isoformas afectadas.
En la retina, la expresión anormal de distrofina es esencialmente responsable de una alteración del electrorretinograma en pacientes.
Estos defectos están directamente relacionados con una alteración de la secuencia de distrofina (que va desde la ausencia hasta una pérdida parcial de la secuencia interna).
Estos conducirán a un colapso más o menos grave de la organización que normalmente forman el complejo macromolecular, alrededor de la distrofina.
Por otro lado, la ausencia de distrofina altera varias vías de señalización intracelular en la fibra del músculo esquelético con los efectos adversos consecuentes de los iones de calcio, las especies reactivas de oxígeno y el óxido nítrico quienes contribuyen cada uno al desarrollo de la distrofia muscular.