
Conocida como RP, es un trastorno genético de los ojos que causa la pérdida de la visión.
Los síntomas incluyen problemas para ver de noche y disminución de la visión periférica (visión lateral).
El inicio de los síntomas es generalmente gradual. A medida que empeora la visión periférica, las personas pueden experimentar una «visión de túnel». La ceguera completa es poco común.
La retinitis pigmentosa generalmente se hereda de los padres de una persona. Mutaciones en uno de más de 50 genes están involucrados. El mecanismo subyacente implica la pérdida progresiva de células fotorreceptoras de la barra en la parte posterior del ojo.
Esto es seguido generalmente por la pérdida de células fotorreceptoras de cono. El diagnóstico es mediante un examen de la retina encontrando depósitos de pigmento oscuro.
Otras pruebas de apoyo pueden incluir un electrorretinograma, pruebas de campo visual o pruebas genéticas.
Actualmente no hay cura para la retinitis pigmentosa. Los esfuerzos para manejar el problema pueden incluir el uso de ayudas para la baja visión, iluminación portátil o un perro guía.
Los suplementos de palmitato de vitamina A pueden ser útiles para retrasar el empeoramiento. Una prótesis visual puede ser una opción en ciertas personas con una enfermedad grave.
Se estima que afecta a 1 de cada 4.000 personas. El inicio es a menudo en la infancia, pero algunos no se ven afectados hasta la edad adulta.
Signos y síntomas
Los síntomas degenerativos retinianos iniciales de la retinitis pigmentosa se caracterizan por disminución de la visión nocturna (nyctalopia) y la pérdida del campo visual periférico medio.
Las células fotorreceptoras de la varilla, que son responsables de la visión con poca luz y están orientadas en la periferia de la retina, son los procesos de la retina afectados primero durante las formas no sindrómicas de esta enfermedad.
El declive visual progresa relativamente rápido hacia el campo periférico lejano, eventualmente extendiéndose al campo visual central a medida que aumenta la visión del túnel.
La agudeza visual y la visión del color pueden verse comprometidas debido a anomalías asociadas en las células fotorreceptoras del cono, que son responsables de la visión del color, la agudeza visual y la vista en el campo visual central.
La progresión de los síntomas de la enfermedad se produce de forma simétrica, con los ojos izquierdo y derecho experimentando síntomas a un ritmo similar.
Una variedad de síntomas indirectos caracterizan a la retinitis pigmentosa junto con los efectos directos de la degeneración del fotorreceptor de la varilla inicial y el descenso posterior de los fotorreceptores del cono.
Fenómenos como la fotofobia, que describe el evento en el que la luz se percibe como un resplandor intenso, y la fotopsia, la presencia de luces parpadeantes o brillantes dentro del campo visual, a menudo se manifiestan durante las últimas etapas de la Retinitis pigmentosa.
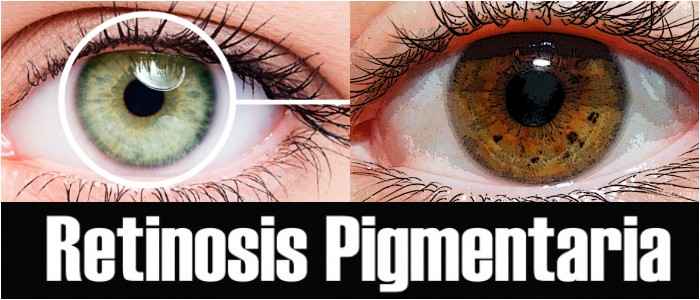
Los hallazgos relacionados con Retinitis pigmentosa a menudo se han caracterizado en el fondo del ojo como la «tríada oftalámica».
Esto incluye el desarrollo de:
- Una apariencia moteada del epitelio pigmentario de la retina (EPR) causado por la formación de espículas óseas.
- Una apariencia cerosa del nervio óptico.
- La atención de los vasos sanguíneos en la retina.
La Retinitis Pigmentosa no sindrómica por lo general presenta una variedad de los siguientes síntomas:
- Ceguera nocturna.
- Visión de túnel (debido a la pérdida de visión periférica).
- Visión de celosía.
- Photopsia (luces parpadeantes/brillantes).
- Fotofobia (aversión a las luces brillantes).
- Desarrollo de espículas óseas en el fondo.
- Ajuste lento de ambientes oscuros a claros y viceversa.
- Visión borrosa.
- Mala separación de color.
- Pérdida de visión central.
- Ceguera eventual.
Causas
La retinitis pigmentosa puede ser:
- No sindrómica, es decir, se produce sola, sin otros hallazgos clínicos.
- Sindrómica, con otros trastornos neurosensoriales, anomalías del desarrollo o hallazgos clínicos complejos.
- Secundaria a otra enfermedades sistémicas.
La retinitis pigmentosa combinada con la sordera (congénita o progresiva) se llama síndrome de Usher.
El síndrome de Alport se asocia con Retinitis pigmentosa y una membrana glomerular-basal anormal que conduce al síndrome nefrótico y se hereda como dominante ligado al cromosoma X.
La retinitis pigmentosa combinada con oftalmoplejía, disfagia, ataxia y defectos de la conducción cardíaca se observa en el síndrome de ADN mitocondrial Síndrome de Kearns-Sayre (también conocido como miopatía de fibra roja rasgada)
En la abetalipoproteinemia se observa retinitis pigmentosa combinada con retraso, neuropatía periférica, glóbulos rojos acantóticos (punteados), ataxia, esteatorrea y ausencia de lipoproteína de muy baja densidad.
La retinitis pigmentosa se observa clínicamente en asociación con otros trastornos genéticos raros (como la distrofia muscular y la enfermedad granulomatosa crónica) como parte del síndrome de McLeod.
Este es un fenotipo recesivo ligado a X caracterizado por una ausencia completa de proteínas de superficie de células XK y, por lo tanto, una expresión marcadamente reducida de todos los antígenos de glóbulos rojos de Kell.
Con fines transfusionales, estos pacientes se consideran completamente incompatibles con todos los donantes normales y K0/K0.
La retinitis pigmentosa asociada con el hipogonadismo y el retraso del desarrollo con un patrón de herencia autosómico recesivo se observa con el síndrome de Bardet-Biedl
Otras afecciones incluyen neurosífilis, toxoplasmosis y enfermedad de Refsum.
Genética
La retinitis pigmentosa (RP) es una de las formas más comunes de degeneración retiniana hereditaria.
Existen múltiples genes que, cuando están mutados, pueden causar el fenotipo de la retinitis pigmentosa.
Los patrones de herencia de Retinitis pigmentosa se han identificado como autosómica dominante, autosómica recesiva, ligada al X y adquirida por vía materna (mitocondrial), y dependen de las mutaciones específicas del gen Retinitis pigmentosa presentes en la generación parental.
En 1989, se identificó una mutación del gen para la rodopsina, un pigmento que desempeña un papel esencial en la cascada de transducción visual que permite la visión en condiciones de poca luz.
El gen de la rodopsina codifica una proteína principal de los segmentos externos de los fotorreceptores.
Las mutaciones en este gen se presentan con mayor frecuencia como mutaciones sin sentido o mal plegamiento de la proteína de la rodopsina, y con mayor frecuencia siguen patrones de herencia autosómica dominante.
Desde el descubrimiento del gen de la rodopsina, se han identificado más de 100 mutaciones del gen RHO (rhodopsin), que representan el 15% de todos los tipos de degeneración retiniana y aproximadamente el 25% de las formas autosómicas dominantes de Retinitis pigmentosa.
Hasta la fecha se han informado hasta 150 mutaciones en el gen opsin asociado con la Retinitis pigmentosa desde que la mutación Pro23 His en el dominio intradiscal de la proteína se informó por primera vez en 1990.
Estas mutaciones se encuentran en todo el gen opsin y se distribuyen a lo largo de los tres dominios de la proteína (los dominios intradiscal, transmembrana y citoplásmico).
Una de las principales causas bioquímicas de la Retinitis pigmentosa en el caso de las mutaciones de la rodopsina es el mal plegamiento de las proteínas y la alteración de las chaperonas moleculares.
Se descubrió que la mutación del codón 23 en el gen de la rodopsina, en el que la prolina se cambia a histidina, representa la fracción más grande de las mutaciones de la rodopsina en los Estados Unidos.
Varios otros estudios han reportado varias mutaciones de codones asociadas con retinitis pigmentosa, incluyendo Thr58Arg, Pro347Leu, Pro347Ser, así como la eliminación de Ile-255.
En 2000, se notificó una mutación rara en el codón 23 que causaba retinosis pigmentaria autosómica dominante, en la que la prolina se transformaba en alanina.
Sin embargo, este estudio demostró que la distrofia retiniana asociada con esta mutación era característicamente leve en presentación y evolución. Además, hubo una mayor conservación en las amplitudes de electrorretinografía que la mutación Pro23His más prevalente.
Se han identificado patrones de herencia autosómica recesiva de Retinitis pigmentosa en al menos 45 genes.
Esto significa que dos individuos no afectados que son portadores de la misma mutación genética inductora de Retinosis pigmentaria en forma dialélica pueden producir descendencia con el fenotipo Retinitis pigmentosa.
Se sabe que una mutación en el gen USH2A causa 10-15% de una forma sindrómica de Retinitis pigmentosa conocida como Síndrome de Usher cuando se hereda de manera autosómica recesiva.
Se sabe que las mutaciones en cuatro factores de empalme pre-ARNm causan retinosis pigmentaria autosómica dominante. Estos son PRPF3 (PRPF3 humano es HPRPF3, también PRP3), PRPF8, PRPF31 y PAP1.
Estos factores se expresan de forma ubicua y se propone que los defectos en un factor ubicuo (una proteína expresada en todas partes) solo deberían causar enfermedad en la retina porque las células fotorreceptoras retinianas tienen un requerimiento mucho mayor para el procesamiento de proteínas (rodopsina) que cualquier otro tipo de célula.
Los patrones de herencia somática o ligada al cromosoma X de Retinitis pigmentosa se identifican actualmente con las mutaciones de seis genes, siendo la más común en loci específicos en los genes RPGR y RP2.
Fisiopatología
Una variedad de defectos de la ruta molecular de la retina se han combinado con múltiples mutaciones conocidas del gen de la Retinosis Pigmentaria.
Las mutaciones en el gen de la rodopsina, que es responsable de la mayoría de los casos de Retinosis pigmentaria autosómica dominante, interrumpen la proteína de la varilla opsina esencial para traducir la luz en señales eléctricas descifrables dentro de la cascada de fototransducción del sistema nervioso central.
Los defectos en la actividad de este receptor acoplado a proteína G se clasifican en distintas clases que dependen de la anomalía de plegamiento específico y los defectos de la ruta molecular resultantes.
La actividad de la proteína mutante de Clase I está comprometida ya que las mutaciones puntuales específicas en la secuencia de aminoácidos que codifican proteínas afectan el transporte de la proteína pigmentaria al segmento externo del ojo, donde se localiza la cascada de fototransducción.
Además, el mal plegamiento de las mutaciones del gen de la rodopsina de clase II altera la conjunción de la proteína con 11-cis-retinal para inducir la formación adecuada de cromóforos.
Los mutantes adicionales en este gen que codifica pigmentos afectan la estabilidad de la proteína, alteran la integridad del ARNm después de la traducción y afectan las velocidades de activación de las proteínas ópticas de transducina y opsina.
Además, los modelos animales sugieren que el epitelio pigmentario de la retina no puede fagocitar los discos del segmento de la varilla exterior que se han desprendido, lo que lleva a una acumulación de restos del segmento de la varilla exterior.
En ratones que son homocigotos recesivos para la mutación de la degeneración de la retina, los fotorreceptores de las varillas dejan de desarrollarse y experimentan degeneración antes de que la maduración celular se complete.
Un defecto en cGMP-phosphodiesterase también se ha documentado; esto conduce a niveles tóxicos de cGMP.
Diagnóstico
Un diagnóstico preciso de retinitis pigmentosa se basa en la documentación de la función de células fotorreceptoras de pérdida progresiva, confirmada por una combinación de pruebas de campo visual y de agudeza visual, fundus e imágenes de coherencia óptica y electroretinografía (ERG),
Las pruebas de campo visual y de agudeza visual miden y comparan el tamaño del campo de visión del paciente y la claridad de su percepción visual con las mediciones visuales estándar asociadas con una visión saludable 20/20.
Las características de diagnóstico clínico indicativas de retinitis pigmentosa incluyen un área visual sustancialmente pequeña y progresivamente decreciente en la prueba de campo visual, y niveles de claridad comprometidos medidos durante la prueba de agudeza visual.
Además, la tomografía óptica, como el fondo y la retina (coherencia óptica), proporcionan otras herramientas de diagnóstico para determinar el diagnóstico de Retinosis pigmentaria.
Fotografiar la parte posterior del ojo dilatado permite confirmar la acumulación de espículas óseas en el fondo, que se presenta durante las últimas etapas de la degeneración retiniana de la Retinosis pigmentaria.
Combinado con imágenes transversales de la tomografía de coherencia óptica, que proporciona pistas sobre:
El grosor de los fotorreceptores, la morfología de la capa de la retina y la fisiología del epitelio pigmentario de la retina, las imágenes del fondo pueden ayudar a determinar el estado de progresión de la Retinosis pigmentaria.
Si bien el campo visual y los resultados de las pruebas de agudeza combinadas con imágenes retinianas respaldan el diagnóstico de retinitis pigmentosa, es necesario realizar pruebas adicionales para confirmar otras características patológicas de esta enfermedad.
La electroretinografía (ERG) confirma el diagnóstico de Retinosis pigmentaria mediante la evaluación de los aspectos funcionales asociados con la degeneración de los fotorreceptores, y puede detectar anomalías fisiológicas antes de la manifestación inicial de los síntomas.
Se aplica una lente de electrodo al ojo a medida que se mide la respuesta de los fotorreceptores a diversos grados de pulsos de luz rápidos.
Los pacientes que exhiben el fenotipo de la retinitis pigmentosa mostrarían una respuesta eléctrica disminuida o retardada en los fotorreceptores de la varilla, así como una respuesta celular del fotorreceptor de cono posiblemente comprometida.
El historial familiar del paciente también se considera al determinar un diagnóstico debido al modo genético de herencia de la retinosis pigmentaria.
Se sabe que al menos 35 genes o loci diferentes causan «retinitis pigmentosa no sindrómica» (Retinitis pigmentosa que no es el resultado de otra enfermedad o parte de un síndrome más amplio).
Las indicaciones del tipo de mutación de Retinosis pigmentaria se pueden determinar a través de pruebas de ADN, que están disponibles sobre una base clínica para:
- RLBP1 (autosómico recesivo, tipo de Bothnia Retinitis pigmentosa).
- RP1 (autosómico dominante, RP1).
- RHO (autosómica dominante, RP4).
- RDS (autosómica dominante, RP7).
- PRPF8 (autosómico dominante, RP13).
- PRPF3 (autosómico dominante, RP18).
- CRB1 (autosómico recesivo, RP12).
- ABCA4 (autosómico recesivo, RP19).
- RPE65 (autosómico recesivo, RP20).
Para todos los demás genes (por ejemplo, DHDDS), las pruebas genéticas moleculares están disponibles solo para investigación.
La retinitis pigmentosa se puede heredar de manera autosómica dominante, autosómica recesiva o ligada a X.
La Retinitis pigmentosa ligada al cromosoma X puede ser recesiva, afectando principalmente solo a los machos, o dominante, afectando tanto a machos como a hembras, aunque los machos generalmente son más levemente afectados.
También se han descrito algunas formas digenéticas (controladas por dos genes) y mitocondriales.
El asesoramiento genético depende de un diagnóstico preciso, la determinación del modo de herencia en cada familia y los resultados de las pruebas genéticas moleculares.
Tratamiento
Actualmente no hay cura para la retinitis pigmentosa, pero la eficacia y seguridad de varios tratamientos prospectivos se están evaluando actualmente.
La eficacia de varios suplementos, como la vitamina A, el ácido docosahexaenoico (DHA) y la luteína, para retrasar la progresión de la enfermedad sigue siendo una opción de tratamiento no resuelta, aunque prospectiva.
Los ensayos clínicos que investigan los dispositivos protésicos ópticos, los mecanismos de terapia génica y los trasplantes de lámina de retina son áreas activas de estudio en la restauración parcial de la visión en pacientes con retinitis pigmentosa.
Los estudios han demostrado el retraso de la degeneración de los fotorreceptores de la varilla mediante la ingesta diaria de 15000 UI (equivalentes a 4,5 mg) de palmitato de vitamina A; por lo tanto, se estanca la progresión de la enfermedad en algunos pacientes.
Investigaciones recientes han demostrado que la administración adecuada de suplementos de vitamina A puede posponer la ceguera hasta por 10 años (al reducir la pérdida del 10% pa al 8,3% anual) en algunos pacientes en ciertas etapas de la enfermedad.
La prótesis de retina Argus se convirtió en el primer tratamiento aprobado para la enfermedad en febrero de 2011, y actualmente está disponible en Alemania, Francia, Italia y el Reino Unido. Los resultados provisionales en 30 pacientes a largo plazo se publicaron en 2012.
El implante de retina Argus II también ha recibido la aprobación del mercado en los EE. UU.
El dispositivo puede ayudar a los adultos con Retinitis pigmentosa que han perdido la capacidad de percibir las formas y el movimiento a ser más móviles y realizar actividades cotidianas.
En junio de 2013, doce hospitales de EE. UU. Anunciaron que pronto aceptarían la consulta de pacientes con Retinitis pigmentosa en preparación para el lanzamiento de Argus II más adelante ese año.
El Alpha-IMS es un implante subretinal que implica la implantación quirúrgica de un pequeño chip de grabación de imágenes debajo de la fóvea óptica.
Las medidas de mejoras visuales de los estudios Alpha-IMS requieren la demostración de la seguridad del dispositivo antes de proceder con los ensayos clínicos y la aprobación del mercado.
El objetivo de los estudios de terapia génica es complementar de forma viral las células de la retina que expresan genes mutantes asociados con el fenotipo de la retinitis pigmentosa con formas sanas del gen.
Por lo tanto, permite la reparación y el funcionamiento adecuado de las células fotorreceptoras retinianas en respuesta a las instrucciones asociadas con el gen sano insertado.
Los ensayos clínicos que investigaron la inserción del gen RPE65 sano en retinas que expresan el fenotipo de retinitis pigmentosa LCA2 midieron modestas mejorías en la visión; sin embargo, la degradación de los fotorreceptores retinianos continuó con la tasa relacionada con la enfermedad.
Probablemente, la terapia génica puede preservar las células retinianas sanas restantes sin reparar la acumulación más temprana de daño en las células fotorreceptoras ya enfermas.
La respuesta a la terapia génica teóricamente beneficiaría a los pacientes jóvenes que muestran la progresión más corta de la disminución de los fotorreceptores; por lo tanto, se correlaciona con una mayor posibilidad de rescate celular a través del gen insertado sano.
Pronóstico
La naturaleza progresiva y la falta de una cura definitiva para la retinitis pigmentosa contribuyen a la perspectiva inevitablemente desalentadora para los pacientes con esta enfermedad.
Si bien la ceguera total es rara, la agudeza visual y el campo visual del paciente continuarán disminuyendo a medida que avance el fotorreceptor de la varilla y la posterior degradación de los fotorreceptores del cono.
Los posibles tratamientos permanecen en las etapas de investigación y ensayo clínico; sin embargo, los estudios de tratamiento sobre la restauración visual en la retinosis pigmentaria son prometedores para el futuro.
Los estudios indican que los niños portadores del genotipo de la enfermedad se benefician del asesoramiento presintomático con el fin de prepararse para las implicaciones físicas y sociales asociadas con la pérdida progresiva de la visión.
Si bien el pronóstico psicológico puede aliviarse ligeramente con el asesoramiento activo, las implicaciones físicas y la progresión de la enfermedad dependen en gran medida de la edad de la manifestación inicial de los síntomas y la tasa de degradación de los fotorreceptores, en lugar del acceso a tratamientos prospectivos.
Las ayudas visuales correctivas y la terapia de la visión personalizada proporcionada por Low Vision Specialists pueden ayudar a los pacientes a corregir pequeñas alteraciones de la agudeza visual y optimizar el campo visual restante.
Los grupos de apoyo, el seguro de la vista y la terapia de estilo de vida son herramientas adicionales útiles para quienes manejan el deterioro visual progresivo.
Epidemiología
La retinitis pigmentosa es la principal causa de ceguera hereditaria, con aproximadamente 1/4,000 personas que experimentan la forma no sindrómica de su enfermedad a lo largo de su vida. Se estima que 1,5 millones de personas en todo el mundo están actualmente afectadas.
La retinitis pigmentosa de aparición temprana se produce en los primeros años de vida y se asocia típicamente con formas de enfermedad sindrómica, mientras que la retinitis pigmentosa de aparición tardía surge desde el inicio hasta la mitad de la edad adulta.
Las formas autosómica dominante y recesiva de retinitis pigmentosa afectan por igual a las poblaciones masculina y femenina.
Sin embargo, la forma menos frecuente de la enfermedad ligada al cromosoma X afecta a los receptores masculinos de la mutación ligada al cromosoma X, mientras que las mujeres suelen permanecer portadoras no afectadas del rasgo Retinitis pigmentosa.
Las formas de la enfermedad ligadas a X se consideran graves y, por lo general, conducen a la ceguera total en etapas posteriores. En raras ocasiones, una forma dominante de la mutación del gen X-linked afectará a hombres y mujeres por igual.
Debido a los patrones de herencia genética de Retinitis pigmentosa, muchas poblaciones aisladas exhiben frecuencias de enfermedad más altas o una mayor prevalencia de una mutación de Retinosis pigmentaria específica.
Las mutaciones preexistentes o emergentes que contribuyen a la degeneración de los fotorreceptores de la varilla en la retinosis pigmentaria se transmiten a través de líneas familiares.
Por lo tanto, permite que ciertos casos de Retinosis Pigmentaria se concentren en regiones geográficas específicas con una historia ancestral de la enfermedad.
Se han realizado varios estudios hereditarios para determinar las tasas de prevalencia variables en Maine (EE. UU.), Birmingham (Inglaterra), Suiza (afecta a 1/7000), Dinamarca (afecta a 1/2500) y Noruega.
Los indios navajos también muestran una tasa elevada de herencia de Retinitis pigmentosa, que se estima afecta a 1 de cada 1878 individuos.
A pesar del aumento de la frecuencia de Retinitis pigmentosa dentro de líneas familiares específicas, la enfermedad se considera no discriminatoria y tiende a afectar por igual a todas las poblaciones mundiales.
Investigación
Los tratamientos futuros pueden incluir trasplantes de retina, implantes retinianos artificiales, terapia génica, células madre, suplementos nutricionales y/o terapias con medicamentos.
2006: investigadores del Reino Unido trasplantaron células madre de ratones que estaban en una etapa avanzada de desarrollo, y ya programadas para desarrollarse en células fotorreceptoras.
En ratones que habían sido inducidos genéticamente para imitar las condiciones humanas de la retinitis pigmentosa y degeneración macular relacionada con la edad.
Estos fotorreceptores desarrollaron e hicieron las conexiones neuronales necesarias a las células nerviosas retinianas del animal, un paso clave en la restauración de la vista.
Anteriormente se creía que la retina madura no tiene capacidad regenerativa. Esta investigación puede llevar en el futuro a utilizar trasplantes en humanos para aliviar la ceguera.
2008: científicos del Instituto de Biociencias de Osaka han identificado una proteína, llamada Pikachurin, que creen que podría conducir a un tratamiento para la retinitis pigmentosa.
2008: se intentó vincular la retinosis pigmentaria con la expresión génica de FAM46A.
2010: una posible terapia genética parece funcionar en ratones.
2012: científicos del Centro médico de la Universidad de Columbia mostraron en un modelo animal que la terapia génica y la terapia de células madre pluripotentes inducidas podrían ser opciones viables para tratar la retinosis pigmentaria en el futuro.
2012: científicos de la Universidad de Miami presentaron datos que muestran la protección de los fotorreceptores en un modelo animal cuando los ojos se inyectan con factor neurotrófico derivado de los astrocitos mesencefálicos.
Investigadores de la Universidad de California en Berkeley lograron restaurar la visión de ratones ciegos mediante la explotación de un «fotoconmutador» que activa las células ganglionares de la retina en animales con células de bastón y cono dañadas.
2015: un estudio de Bakondi et al. en el Centro Médico Cedars-Sinai demostró que CRISPR/Cas9 puede usarse para tratar ratas con la forma autosómica dominante de retinitis pigmentosa.
2016: RetroSense Therapeutics intentó inyectar virus con ADN de algas sensibles a la luz en los ojos de varias personas ciegas (que tienen retinitis pigmentosa). Si tiene éxito, podrán ver en blanco y negro.


