
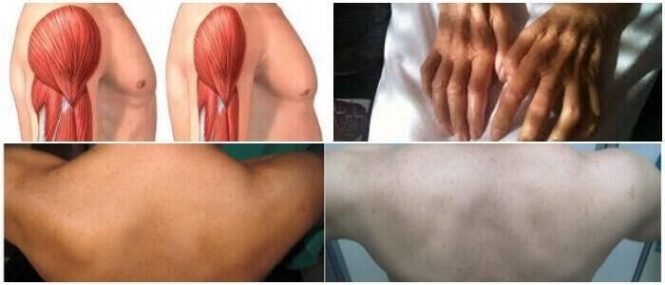
Es un desgaste progresivo de los tejidos musculares.
El dolor muscular también es un síntoma. Puede ocurrir en hombres de mediana edad con diabetes tipo 2. También ocurre con la enfermedad de la neurona motora.
Neuropatía diabética proximal
La neuropatía diabética proximal, más comúnmente conocida como amiotrofia diabética, es un trastorno nervioso que se produce como una complicación de la diabetes mellitus. Puede afectar los muslos, las caderas, las nalgas o la parte inferior de las piernas.
La neuropatía diabética proximal es una enfermedad de los nervios periféricos (neuropatía diabética) caracterizada por desgaste muscular o debilidad, dolor o cambios en la sensación/entumecimiento de la pierna.
La neuropatía diabética es una complicación infrecuente de la diabetes. Es un tipo de plexopatía lumbosacra, o afección adversa que afecta el plexo lumbosacro.
Hay varias formas en que la diabetes daña los nervios, todas las cuales parecen estar relacionadas con el aumento de los niveles de azúcar en la sangre durante un período prolongado. La neuropatía diabética proximal es uno de los cuatro tipos de neuropatía diabética.
La neuropatía diabética proximal puede ocurrir en pacientes con diabetes mellitus tipo 2 y tipo 1, sin embargo, se encuentra con mayor frecuencia en diabéticos tipo 2. La neuropatía proximal es el segundo tipo más común de neuropatía diabética y puede resolverse con tiempo y tratamiento.
Signos y síntomas
Los signos y síntomas de la neuropatía diabética proximal dependen de la región del plexo afectada. El primer síntoma suele ser dolor en las nalgas, caderas, muslos o piernas. Este dolor generalmente afecta un lado del cuerpo y puede comenzar gradualmente o aparecer de repente.
Esto a menudo va seguido de debilidad variable en los músculos proximales de las extremidades inferiores. Estos síntomas, aunque a menudo comienzan en un lado, también pueden diseminarse a ambos lados.
La debilidad en la neuropatía diabética proximal es causada por la denervación de los músculos específicos inervados por las regiones del plexo afectadas y por lo tanto estos músculos pueden comenzar a exhibir fasciculaciones.
Tenga en cuenta que la amiotrofia diabética es una condición causada por la diabetes mellitus, pero separada de la condición más común de polineuropatía.
Causas
Primero se pensó que el daño nervioso asociado con la enfermedad era causado por cambios metabólicos como la enfermedad de los microvasos endoneurales, que es la degeneración de los pericitos debido a la hiperglucemia y la reproducción de las membranas basales cuando los pericitos ya no regulan su ciclo celular.
El tamaño disminuido del lumen más la ausencia del pericito, que regula el flujo sanguíneo capilar y la fagocitosis de los restos celulares, conduce a la isquemia. Las biopsias de los nervios han cambiado la vista hacia un mecanismo inmune que causa Micro Vasculitis, que eventualmente podría conducir a la isquemia.
Los tratamientos experimentales que usan proteínas inmunosupresoras han proporcionado evidencia corroborativa adicional a la teoría del mecanismo inmune.
Aunque esta enfermedad ocurre en pacientes sin diabetes, la prevalencia es mucho mayor en diabéticos, lo que indica que, aunque la hiperglucemia no causa directamente el daño a los nervios, puede desempeñar un papel.
Diagnóstico
Con frecuencia, se sospecha que los pacientes con diabetes y dolor y debilidad proximales (cadera, muslo) tienen amiotrofia diabética.
El diagnóstico más definitivo se hace comúnmente con estudios de electrodiagnóstico que incluyen estudios de conducción nerviosa y electromiograma.
La amiotrofia diabética a menudo es un diagnóstico de exclusión en pacientes diabéticos con una plexopatía lumbosacra para la que no se puede determinar ninguna otra causa de la plexopatía lumbosacra.
Tratamiento
El manejo adecuado de la diabetes mellitus puede prevenir que ocurra una neuropatía diabética proximal.
Se cree que la incidencia de la incidencia de neuropatía diabética proximal se correlaciona con el control de la glucosa en sangre en diabéticos, y es probable que sea reversible con un mejor control.
La medicación ayuda a reducir el dolor involucrado en la neuropatía diabética proximal. La mayoría de los pacientes toman medicamentos orales recetados por un médico.
Los tipos comunes de medicamentos utilizados para tratar la amiotrofia diabética incluyen anticonvulsivos (por ejemplo, gabapentina, pregabalina) así como medicamentos opioides, aunque esta última categoría no está óptimamente indicada para el dolor neuropático.
Amiotrofia monomelica
La amiotrofia monomélica (MMA), también conocida como enfermedad de Hirayama y atrofia muscular espinal juvenil no progresiva, es una enfermedad de la neurona motora que fue descrita por primera vez por Keizo Hirayama en 1959; Mandavilli Gourie-Devi (et al) introdujo el término «monotélico amiotrofia» en 1984.
La enfermedad (trastorno) principalmente (pero no exclusivamente) afecta a hombres jóvenes (de 15 a 25 años de edad) en Asia, con la mayoría de los casos estudiados en India y Japón.
A partir de 2011, se habían escrito alrededor de 200 casos, comenzando con los 38 pacientes en el estudio de Hirayama de 1959. Tanto los nombres para el desorden como sus posibles causas han estado evolucionando desde que se informaron por primera vez.
Signos y síntomas
Los síntomas incluyen una aparición lenta de atrofia muscular, que se estabiliza en una meseta después de dos a cinco años a partir de los cuales no mejora ni empeora. No hay dolor o pérdida sensorial asociada con amiotrofia monomélica y las fasciculaciones (contracciones musculares involuntarias) son raras.
Existe un debate sobre si esta afección representa una forma focal de degeneración primaria de la neurona motora inferior (es decir, una forma focal de atrofia muscular espinal) o una consecuencia local de la compresión crónica a partir de una expansión dural en la columna cervical.
En las primeras etapas de la enfermedad, la amiotrofia monomérica puede confundirse con el síndrome del túnel carpiano avanzado y con las etapas iniciales de la esclerosis lateral amiotrófica (ELA).
Los síntomas difieren algo. El dolor y el hormigueo en la mano están típicamente presentes en síndrome del túnel carpiano y ausentes de la amiotrofia monomélica; la pérdida de función se presenta de manera diferente; con cuidadoso estudio electrofisiológico y exámenes neurológicos, los dos se distinguen.
En la esclerosis lateral amiotrófica temprana frente a la amiotrofia monomélica, la presentación es similar.
En la esclerosis lateral amiotrófica, los síntomas de la mano son más comunes proximal y distal que en la amiotrofia monomérica, principalmente distal, y con fasciculaciones de la esclerosis lateral amiotrófica (espasmos) a menudo están presentes en las extremidades superiores, pero raramente en la amiotrofia monomélica.
La amiotrofia monomélica generalmente se elimina de la consideración si la discapacidad se expresa en más de una extremidad o en extremidades inferiores (piernas), pero la ausencia sintomática puede no descartar la esclerosis lateral amiotrófica durante tres a cinco años después del inicio inicial.
Textos electrofisiológicos y pruebas reflejas tienden a producir resultados diferentes, pero la interpretación es a veces subjetiva.
Causas
La causa exacta de la amiotrofia monomelica no se entiende bien. Se cree que la enfermedad puede ocurrir cuando el material que rodea la médula espinal (saco tecal o saco dural) cambia de posición. Esto puede ser causado por movimientos repetidos hacia abajo (flexión) del cuello.
El cambio de posición del saco dural puede causar presión sobre la médula espinal. Esto puede afectar la capacidad de las señales que se envían desde el cerebro a los músculos del brazo. Esto podría causar los signos y síntomas de la amiotrofia monomélica.
Sin embargo, no se ha confirmado que la presión sobre la médula espinal explica por qué algunas personas desarrollan amiotrofia monomérica. Otras causas posibles incluyen disfunción del sistema inmune o una infección.
Herencia
La amiotrofia monomelica generalmente no se cree que sea hereditaria; se ha descrito un vínculo familiar solo en un porcentaje menor de casos.
La amiotrofia monomelica no se cree que sea causada por cambios en un gen específico. La mayoría de las personas que tienen amiotrofia monomélica son las únicas personas con la enfermedad en la familia. En algunos casos, las personas con amiotrofia monomérica informaron tener otros familiares con la enfermedad.
En un caso, se informó que dos gemelos idénticos desarrollaron la enfermedad. Esto hace que los investigadores piensen que puede haber factores genéticos que predisponen a las personas a desarrollar amiotrofia monomérica.
Sin embargo, no todos los que tienen estos factores genéticos necesariamente desarrollarán amiotrofia monomélica. En cambio, es probablemente una combinación de factores genéticos y ambientales que hacen que las personas desarrollen amiotrofia monomérica.
Diagnóstico
La amiotrofia monomelica se sospecha cuando un doctor observa los signos y síntomas de la enfermedad, como la debilidad muscular en un brazo, solo que comienza durante la adolescencia o la adultez temprana. El diagnóstico se puede confirmar con estudios de imágenes y pruebas de laboratorio.
Los estudios de imágenes que pueden ayudar a confirmar un diagnóstico de amiotrofia monomélica incluyen imágenes por resonancia magnética o tomografías computarizadas. Estos estudios de imágenes pueden mostrar signos de compresión de porciones de la médula espinal.
Las pruebas de laboratorio pueden incluir un electromiógrafo que muestra una respuesta reducida en los nervios que llevan señales a los músculos del brazo. Deben descartarse otras posibles causas de debilidad muscular como trauma o lesión para confirmar el diagnóstico de la amiotrofia monomélica.
No hay cura para la amiotrofia monomelica. La investigación ha encontrado formas de manejar mejor la enfermedad, incluido el uso de un collarín cervical, ejercicios de fortalecimiento muscular y capacitación en coordinación manual.
Debido a que las intervenciones tempranas son aparentemente más útiles, otros desenlaces y causas, como el túnel carpiano, la esclerosis lateral amiotrófica, los tumores y los traumatismos, deben eliminarse tan pronto como sea posible.
Tratamiento
Desafortunadamente, no hay cura para la amiotrofia monomelica. Sin embargo, existen opciones de tratamiento que pueden ayudar a controlar los síntomas de la enfermedad y ralentizar la progresión de la debilidad muscular.
Si los médicos piensan que la amiotrofia monomélica es causada por la compresión de la médula espinal, pueden recomendar el uso de un aparato ortopédico que se puede usar alrededor del cuello para evitar el movimiento hacia abajo (flexión) del cuello.
Otras opciones de tratamiento incluyen ejercicios de fortalecimiento muscular y terapias para mejorar la coordinación de la mano. La cirugía para el tratamiento de la amiotrofia monomérica se debate, ya que existen beneficios y riesgos asociados con la cirugía.
Las personas que son diagnosticadas con amiotrofia monomérica probablemente serán recomendadas para ver a un especialista neuromuscular que pueda vigilar la progresión de la enfermedad.
Pronóstico
La perspectiva a largo plazo para las personas con amiotrofia monomélica es generalmente buena. Aunque los síntomas de la enfermedad pueden progresar durante algunos años después de que comienza la debilidad muscular inicial, los síntomas generalmente se estabilizan.
La debilidad muscular generalmente afecta a un brazo y no se asocia con dolor u otros síntomas.
Algunas personas con amiotrofia monomélica tienen pérdida de la función de una mano. Esto puede causar dificultades en el cuidado de uno mismo, el trabajo y las situaciones sociales. Las sesiones con terapeutas ocupacionales o trabajadores sociales pueden ayudar a superar las dificultades asociadas con la debilidad muscular.
La esclerosis lateral amiotrófica
Algunos también usan el término enfermedad de la neurona motora para un grupo de condiciones de las cuales la esclerosis lateral amiotrófica es la más común.
La esclerosis lateral amiotrófica se caracteriza por rigidez muscular, espasmos musculares y empeoramiento gradual de la debilidad debido a la disminución de tamaño de los músculos. Esto resulta en dificultad para hablar, tragar y finalmente respirar.
La causa no se conoce en 90% a 95% de los casos. El 5-10% restante de los casos se hereda de los padres de una persona. Aproximadamente la mitad de estos casos genéticos se deben a uno de dos genes específicos.
El mecanismo subyacente implica daño a las neuronas motoras superiores e inferiores. El diagnóstico se basa en los signos y síntomas de una persona, y se realizan pruebas para descartar otras posibles causas.
Un medicamento llamado riluzol puede prolongar la vida en aproximadamente dos o tres meses. La ventilación no invasiva puede dar como resultado una mejor calidad y duración de la vida.
La enfermedad puede afectar a personas de cualquier edad, pero generalmente comienza alrededor de la edad de 60 años y en casos heredados alrededor de los 50 años. La supervivencia promedio desde el inicio hasta la muerte es de dos a cuatro años. Alrededor del 10% sobrevive más de 10 años.
Signos y síntomas
El trastorno causa debilidad muscular, atrofia y espasmos musculares en todo el cuerpo debido a la degeneración del motor superior y las neuronas motoras inferiores.
Las personas afectadas por el trastorno pueden, en última instancia, perder la capacidad de iniciar y controlar todo el movimiento voluntario, aunque la función de la vejiga, el intestino y los músculos responsables del movimiento ocular generalmente se evitan hasta las etapas finales del trastorno.
La disfunción cognitiva o conductual está presente en 30-50% de las personas con esclerosis lateral amiotrófica. Alrededor de la mitad de las personas con esclerosis lateral amiotrófica experimentarán cambios leves en la cognición y el comportamiento, y 10-15% mostrarán signos de demencia frontotemporal.
La repetición de frases o gestos, la apatía y la pérdida de la inhibición con frecuencia se informan características del comportamiento de la esclerosis lateral amiotrófica.
La disfunción del lenguaje, la disfunción ejecutiva y los problemas con la cognición social y la memoria verbal son los síntomas cognitivos más frecuentes en la esclerosis lateral amiotrófica; un metanálisis no encontró relación entre la disfunción y la gravedad de la enfermedad.
Aproximadamente la mitad de las personas con esclerosis lateral amiotrófica experimentan labilidad emocional, en la cual lloran o ríen sin razón.
Los nervios sensoriales y el sistema nervioso autónomo generalmente no se ven afectados, lo que significa que la mayoría de las personas con esclerosis lateral amiotrófica mantienen la audición, la vista, el tacto, el olfato y el gusto.
Síntomas iniciales
El inicio de la esclerosis lateral amiotrófica puede ser tan sutil que los síntomas se pasan por alto. Los primeros síntomas de la esclerosis lateral amiotrófica son debilidad muscular o atrofia muscular.
En la esclerosis lateral amiotrófica de inicio de extremidades, las personas primero experimentan incomodidad al caminar o correr o incluso al tropezarse, y esto se caracteriza por caminar con un «pie caído» que se arrastra suavemente por el suelo.
En la esclerosis lateral amiotrófica de inicio bulbar, los síntomas iniciales serán principalmente de dificultad para hablar con claridad o para tragar.
El habla puede volverse difícil, de carácter nasal o más silenciosa. Puede haber dificultad para tragar y pérdida de la movilidad de la lengua.
Una menor proporción de personas experimenta una esclerosis lateral amiotrófica de «inicio respiratorio», donde los músculos intercostales que soportan la respiración se afectan primero.
Los síntomas de afectación de la neurona motora superior incluyen músculos tensos y rígidos (espasticidad) y reflejos exagerados (hiperreflexia) que incluyen un reflejo nauseoso hiperactivo.
Un reflejo anormal comúnmente llamado signo de Babinski también indica daño de la neurona motora superior.
Progresión
Aunque el orden y la tasa de síntomas varían de persona a persona, la enfermedad eventualmente se propaga a regiones no afectadas y las regiones afectadas se vuelven más afectadas.
La mayoría de las personas eventualmente no pueden caminar o usar sus manos y brazos, pierden la capacidad de hablar y tragar los alimentos y su propia saliva, y comienzan a perder la capacidad de toser y respirar por sí mismos.
La tasa de progresión se puede medir utilizando una medida de resultado llamada «Escala de calificación funcional de la esclerosis lateral amiotrófica revisada».
Un instrumento de 12 ítems administrado como entrevista clínica o cuestionario autoinformado que arroja un puntaje entre 48 (función normal) y 0 (discapacidad grave); es la medida de resultado más comúnmente utilizada en los ensayos clínicos y es utilizada por los médicos para rastrear la progresión de la enfermedad.
Aunque el grado de variabilidad es alto y un pequeño porcentaje de personas tiene un trastorno mucho más lento, en promedio, las personas con esclerosis lateral amiotrófica pierden aproximadamente 0.9 puntos de escala de calificación funcional por mes.
Etapas tardías
La dificultad para masticar y tragar hace que comer sea muy difícil y aumenta el riesgo de asfixia o aspiración de alimentos hacia los pulmones.
En etapas posteriores del trastorno, puede desarrollar neumonía por aspiración y mantener un peso saludable puede convertirse en un problema importante que puede requerir la inserción de un tubo de alimentación.
A medida que el diafragma y los músculos intercostales de la caja torácica que soportan la respiración se debilitan, las medidas de la función pulmonar, como la capacidad vital y la presión inspiratoria, disminuyen.
En la esclerosis lateral amiotrófica de inicio respiratorio, esto puede ocurrir antes de que la debilidad significativa de las extremidades sea evidente. La mayoría de las personas con esclerosis lateral amiotrófica mueren por insuficiencia respiratoria o neumonía.
Causas
La característica definitoria de la esclerosis lateral amiotrófica es la muerte de las neuronas motoras superiores e inferiores en la corteza motora del cerebro, el tronco encefálico y la médula espinal.
Antes de su destrucción, las neuronas motoras desarrollan inclusiones ricas en proteínas en sus cuerpos celulares y axones. Esto puede deberse en parte a defectos en la degradación de la proteína.
Estas inclusiones a menudo contienen ubiquitina, y generalmente incorporan una de las proteínas asociadas a la esclerosis lateral amiotrófica: SOD1, proteína de unión al ADN TAR (TDP-43 o TARDBP) o FUS. El SOD1 mutante también puede contribuir a la muerte de las neuronas motoras mediante la generación de radicales libres.
La excitotoxicidad, o muerte celular causada por niveles elevados de calcio intracelular causados por la actividad excesiva de neurotransmisores excitatorios, puede ser un mecanismo de la esclerosis lateral amiotrófica.
Este concepto ha sido respaldado por un aumento del glutamato y del ARN transportador de glutamato disfuncional en el líquido cefalorraquídeo de las personas con esclerosis lateral amiotrófica.
Esto es apoyado además por el único tratamiento efectivo que es un fármaco anti-glutaminérgico (Riluzol), así como la poca capacidad de amortiguar el calcio en las neuronas motoras en relación con otras neuronas.
La acumulación de neurofilamentos en los axones se ha observado en casos esporádicos de esclerosis lateral amiotrófica, así como en pacientes con SOD1, posiblemente interfiriendo con el transporte axonal, lo que lleva a la muerte celular por toxicidad de SOD1.
Genética
Alrededor del 5-10% de los casos son heredados directamente de los padres de una persona. En general, los parientes de primer grado de un individuo con esclerosis lateral amiotrófica tienen un riesgo del 1% de desarrollar esclerosis lateral amiotrófica.
Un defecto en el cromosoma 21, que codifica para la superóxido dismutasa, se asocia con alrededor del 20% de los casos familiares de esclerosis lateral amiotrófica, o alrededor del 2% de los casos de esclerosis lateral amiotrófica en general.
Lesión craneal
Aunque la lesión cerebral traumática de moderada a severa es un riesgo de esclerosis lateral amiotrófica, si la lesión cerebral traumática leve aumenta las tasas no está claro.
La esclerosis lateral amiotrófica también puede ocurrir más a menudo entre los veteranos del ejército de EE. UU., Pero se desconoce el motivo.
Otros factores
Cuando no hay antecedentes familiares de la enfermedad, es alrededor del 90% de los casos, no se conoce ninguna causa. Las posibles asociaciones para las que la evidencia no es concluyente incluyen el servicio militar y el tabaquismo.
Aunque los estudios sobre la historia militar y la frecuencia de la esclerosis lateral amiotrófica son inconsistentes, hay evidencia débil de una correlación positiva.
Varios factores propuestos incluyen la exposición a toxinas ambientales (inferidas a partir de estudios de despliegue geográfico), así como el consumo de alcohol y tabaco durante el servicio militar.
Otros factores de riesgo potenciales permanecen sin confirmar, incluida la exposición a sustancias químicas, la exposición al campo electromagnético, la ocupación, los traumatismos físicos y las descargas eléctricas.
Diagnóstico
Se están estudiando varios biomarcadores para la afección, pero hasta ahora no son de uso médico general.
Las enfermedades infecciosas virales como el virus de la inmunodeficiencia humana (VIH), el virus linfotrópico T humano, la enfermedad de Lyme, la sífilis y la encefalitis transmitida por garrapatas pueden en algunos casos causar síntomas similares a la esclerosis lateral amiotrófica.
Los trastornos neurológicos tales como esclerosis múltiple, síndrome de post-polio, neuropatía motora multifocal, polineuropatía desmielinizante inflamatoria crónica, atrofia muscular espinal, y la atrofia muscular bulbar también pueden imitar ciertos aspectos de la enfermedad y debe ser considerado.
La esclerosis lateral amiotrófica debe diferenciarse de los «síndromes mímicos esclerosos amiotróficos laterales», que son trastornos no relacionados que pueden tener una presentación y características clínicas similares a la esclerosis lateral amiotrófica o sus variantes.
Debido al pronóstico de este diagnóstico y la variedad de enfermedades o trastornos que pueden parecerse a la esclerosis lateral amiotrófica en las primeras etapas de la enfermedad, las personas con síntomas amiotróficos de esclerosis lateral siempre deben obtener una opinión neurológica especializada para descartar diagnósticos alternativos.
El síndrome miasténico, también conocido como síndrome de Lambert-Eaton, puede simular esclerosis lateral amiotrófica, y su presentación inicial puede ser similar a la de la miastenia gravis, una enfermedad autoinmune tratable que a veces se confunde con esclerosis lateral amiotrófica.
El síndrome de fasciculación benigna es otra afección que imita algunos de los primeros síntomas de la esclerosis lateral amiotrófica, pero se acompaña de lecturas de electromiografía normales y no presenta una discapacidad importante.
La mayoría de los casos de esclerosis lateral amiotrófica, sin embargo, se diagnostican correctamente.
Tratamiento
Esta atención de apoyo es mejor provista por equipos multidisciplinarios de profesionales de la salud que trabajan con la persona y sus cuidadores para mantenerlos tan móviles y cómodos como sea posible.
Medicamentos
Se ha encontrado que el riluzol prolonga modestamente la supervivencia en aproximadamente 2-3 meses. Puede tener un mayor beneficio de supervivencia para aquellos con un inicio bulbar.
Está aprobado por la Administración de Alimentos y Medicamentos de los Estados Unidos y recomendado por el Instituto Nacional de Excelencia en Salud y Atención en Inglaterra y Gales.
El riluzol no revierte el daño ya hecho a las neuronas motoras, pero afecta a las neuronas al reducir su actividad al bloquear la entrada de Na+ a las neuronas, bloqueando así la liberación de los químicos que causan la actividad de las neuronas motoras.
La reducción de la actividad evita la destrucción del músculo neuronal y, por lo tanto, la droga puede actuar como una sustancia química protectora. La función de este medicamento depende de la cantidad tomada en un momento dado.
Cuanto mayor sea la concentración, mejor protegerá la droga a las neuronas de la ruina. La dosis recomendada de riluzol es de 50 mg, dos veces al día para personas con esclerosis lateral amiotrófica conocida durante más de cinco años.
Una cantidad de efectos secundarios son causados por el medicamento, incluida la sensación de debilidad en los músculos, pero esto es normal debido a la función del medicamento.
Los estudios han demostrado que es poco probable que las personas que toman el medicamento dejen de responder a él o desarrollen síntomas que puedan hacer que la actividad de las neuronas vuelva a aumentar, lo que lo convierte en un fármaco eficaz para prolongar la supervivencia.
En 2015, se aprobó la edaravona en Japón para el tratamiento de la esclerosis lateral amiotrófica después de estudiar cómo y si funciona en 137 personas con esclerosis lateral amiotrófica y ha obtenido el estado de fármaco huérfano en la Unión Europea y Estados Unidos.
También hay medicamentos disponibles para ayudar a las personas con dolor, como medicamentos no esteroideos y antiinflamatorios y opioides, depresión, alteraciones del sueño, disfagia y estreñimiento.
El baclofeno y el diazepam a menudo se recetan para controlar la espasticidad causada por la esclerosis lateral amiotrófica, y se puede recetar trihexifenidilo, amitriptilina o, más comúnmente, glicopirrolato cuando las personas con esclerosis lateral amiotrófica comienzan a tener problemas para tragar su saliva.
No hay evidencia de que los medicamentos sean efectivos para reducir los calambres musculares experimentados por personas con esclerosis lateral amiotrófica.
Respiración de soporte
La insuficiencia respiratoria es la causa más común de muerte en personas con esclerosis lateral amiotrófica y es el síntoma más prominente, en segundo lugar a la destrucción de las neuronas motoras y el debilitamiento del músculo.
Cuando los músculos que ayudan a respirar se debilitan, comienzan a aparecer varios síntomas, como dificultad para respirar al realizar actividad física o hablar, fatiga, dolores de cabeza matinales, falta de concentración y depresión.
La ventilación con coraza bifásica tiene la ventaja adicional de ser capaz de ayudar a despejar las secreciones mediante el uso de oscilaciones de alta frecuencia seguidas de varias respiraciones espiratorias positivas.
Las personas pueden eventualmente considerar formas de ventilación mecánica (respiradores) en los que una máquina infla y desinfla los pulmones.
Para ser eficaz, esto puede requerir un tubo que pasa desde la nariz o la boca hasta la tráquea y para el uso a largo plazo, una operación como una traqueotomía, en la que se inserta un tubo de respiración de plástico directamente en la tráquea de la persona en una abertura en el cuello.
Las personas y sus familias deben considerar varios factores al momento de decidir si usar una de estas opciones y cuándo hacerlo. Los dispositivos de ventilación difieren en su efecto sobre la calidad de vida y el costo para la persona.
Aunque el soporte ventilatorio puede aliviar los problemas respiratorios y prolongar la supervivencia, no afecta la progresión de la esclerosis lateral amiotrófica.
Las personas necesitan estar completamente informadas sobre estas consideraciones y los efectos a largo plazo de la vida sin movimiento antes de tomar decisiones sobre el soporte de ventilación y tener discusiones profundas sobre la calidad de vida.
Algunas personas sometidas a una traqueotomía a largo plazo con ventilación con presión positiva intermitente con manguitos desinflados o tubos de traqueostomía sin manguito (ventilación con fugas) pueden hablar, siempre que sus músculos bulbares sean lo suficientemente fuertes, aunque en todos los casos se perderá el habla a medida que progresa la enfermedad.
Esta técnica preserva el habla en algunas personas con ventilación mecánica a largo plazo. Otras personas pueden usar una válvula de habla, como una válvula de habla Passey-Muir, con la asistencia y orientación de un patólogo del habla y el lenguaje.
Las máquinas de ventilación externa que utilizan el modo de ventilación de la presión positiva de dos niveles en las vías respiratorias se usan con frecuencia para tratar la insuficiencia respiratoria por la noche y más tarde durante el día.
El uso de la presión positiva en dos niveles (más comúnmente conocida como ventilación no invasiva) ha demostrado que prolonga la supervivencia y ralentiza la progresión de la capacidad vital forzada.
Pero mucho antes de que la presión positiva en las vías respiratorias deje de ser efectiva, las personas deben decidir si realizar una traqueotomía y ventilación mecánica a largo plazo. En este punto, algunas personas eligen cuidados paliativos para enfermos terminales.
Terapia
La fisioterapia juega un papel importante en la rehabilitación de individuos con esclerosis lateral amiotrófica.
Específicamente, los terapeutas físicos, ocupacionales y del habla pueden establecer objetivos y promover beneficios para las personas con esclerosis lateral amiotrófica al retrasar la pérdida de fuerza, mantener la resistencia, limitar el dolor, mejorar el habla y la deglución, prevenir complicaciones y promover la independencia funcional.
La terapia ocupacional y el equipo especial como la tecnología de asistencia también pueden mejorar la independencia y la seguridad de las personas a lo largo de la esclerosis lateral amiotrófica.
El ejercicio aeróbico suave y de bajo impacto, como realizar actividades de la vida diaria, caminar, nadar y andar en bicicleta estática puede fortalecer los músculos no afectados, mejorar la salud cardiovascular y ayudar a las personas a combatir la fatiga y la depresión.
La amplitud de movimiento y los ejercicios de estiramiento pueden ayudar a prevenir la espasticidad dolorosa y el acortamiento (contractura) de los músculos.
Los terapeutas físicos y ocupacionales pueden recomendar ejercicios que brinden estos beneficios sin sobrecargar los músculos, ya que el agotamiento muscular puede empeorar los síntomas asociados con la esclerosis lateral amiotrófica, en lugar de brindar ayuda a las personas con esclerosis lateral amiotrófica.
Pueden sugerir dispositivos como rampas, aparatos ortopédicos, andadores, equipos de baño (sillas de ducha, elevadores de inodoros, etc.) y sillas de ruedas que ayudan a las personas a mantenerse móviles.
Los terapeutas ocupacionales pueden proporcionar o recomendar equipos y adaptaciones para permitir que la gente con esclerosis lateral amiotrófica conserve tanta seguridad e independencia en las actividades de la vida diaria como sea posible.
Las personas con esclerosis lateral amiotrófica que tienen dificultades para hablar pueden beneficiarse al trabajar con un patólogo del habla y el lenguaje. Estos profesionales de la salud pueden enseñar a las personas estrategias de adaptación, como técnicas para ayudarlas a hablar más fuerte y con mayor claridad.
A medida que progresa la esclerosis lateral amiotrófica, los patólogos del habla y el lenguaje pueden recomendar el uso de comunicación aumentativa como amplificadores de voz, dispositivos generadores de voz o técnicas de comunicación de baja tecnología como punteros láser montados en la cabeza o señales de sí y no.
Nutrición
Las personas con esclerosis lateral amiotrófica y cuidadores pueden aprender de los dietistas cómo planificar y preparar numerosas comidas pequeñas a lo largo del día que proporcionan suficientes calorías, fibra y líquidos, y cómo evitar los alimentos que son difíciles de tragar.
Proporcionar comidas con vitamina E y tomar suplementos de vitamina E ha demostrado que ralentiza la progresión de la esclerosis lateral amiotrófica. Las personas pueden comenzar a usar dispositivos de succión para eliminar el exceso de líquidos o saliva y evitar la asfixia.
Los terapeutas ocupacionales pueden ayudar con las recomendaciones de equipos adaptativos para facilitar la tarea física de la autoalimentación. Los patólogos del habla y del lenguaje hacen recomendaciones de elección de alimentos que son más conducentes a sus deficiencias y capacidades únicas.
Cuando las personas con esclerosis lateral amiotrófica ya no pueden alimentarse lo suficiente, los médicos pueden aconsejar insertar un tubo de alimentación en el estómago.
Cuidado al final de la vida
Los trabajadores sociales y las enfermeras de cuidados domiciliarios y de cuidados paliativos ayudan a las personas con esclerosis lateral amiotrófica, sus familias y cuidadores a enfrentar los desafíos médicos, emocionales y financieros, particularmente durante las etapas finales de la enfermedad.
Los trabajadores sociales brindan apoyo, como asistencia para obtener ayuda financiera, organización de un poder notarial duradero, preparación de un testamento en vida y búsqueda de grupos de apoyo para pacientes y cuidadores.
Las enfermeras a domicilio están disponibles no solo para proporcionar atención médica, sino también para enseñar a los cuidadores sobre tareas tales como mantener los respiradores, dar de comer y mover a las personas para evitar problemas y contracturas dolorosas en la piel.
Las enfermeras de hospicio trabajan en consulta con los médicos para garantizar la medicación adecuada, el control del dolor y otros cuidados que afectan la calidad de vida de las personas con esclerosis lateral amiotrófica que desean permanecer en sus hogares.
El equipo de hospicio en el hogar también puede aconsejar a las personas con esclerosis lateral amiotrófica y a los cuidadores acerca de los problemas al final de la vida.