Es un fluido secretado por las células de los alvéolos (los pequeños sacos de aire en los pulmones) que sirve para reducir la tensión superficial de los fluidos pulmonares.
El surfactante contribuye a las propiedades elásticas del tejido pulmonar, evitando que los alvéolos colapsen.
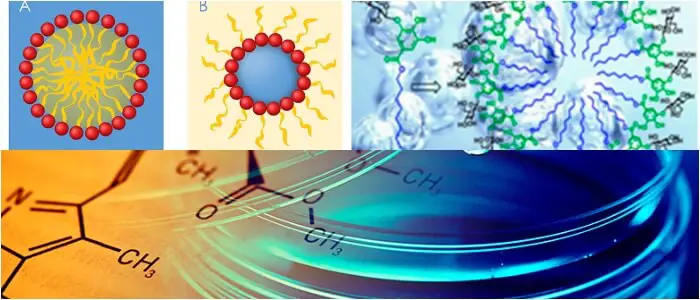
El surfactante pulmonar es un complejo de lipoproteína de superficie activa (fosfolipoproteína) formado por células alveolares tipo II. Las proteínas y los lípidos que componen el tensoactivo tienen regiones hidrofílicas e hidrofóbicas.
Adsorbiendo a la interfase aire-agua de los alvéolos, con grupos hidrofílicos en el agua y las colas hidrófobas mirando hacia el aire, el principal componente lipídico del surfactante, dipalmitoilfosfatidilcolina (DPPC), reduce la tensión superficial.
Como medicamento, el surfactante pulmonar figura en la Lista Modelo de Medicamentos Esenciales de la OMS, los medicamentos más importantes que se necesitan en un sistema de salud básico.
Función
- Para aumentar el cumplimiento pulmonar.
- Para prevenir la atelectasia (colapso del pulmón) al final de la espiración.
- Para facilitar el reclutamiento de las vías respiratorias colapsadas.
Los alvéolos se pueden comparar con el gas en el agua, ya que los alvéolos están húmedos y rodean un espacio aéreo central.
Conformidad
El cumplimiento es la capacidad de expansión de los pulmones y el tórax. La conformidad pulmonar se define como el cambio de volumen por unidad de cambio de presión a través del pulmón.
Esta diferencia en los volúmenes de inflado y deflación a una presión dada se denomina histéresis y se debe a la tensión superficial del aire-agua que se produce al comienzo de la inflación.
Sin embargo, el tensoactivo disminuye la tensión de la superficie alveolar, como se observa en los casos de bebés prematuros que padecen el síndrome de dificultad respiratoria infantil.
La tensión superficial normal para el agua es de 70dyn/cm (70mN/m) y en los pulmones es de 25dyn/cm (25mN/m); sin embargo, al final de la espiración, las moléculas de fosfolípidos tensoactivos comprimidos disminuyen la tensión superficial a niveles muy bajos, cercanos a cero.
Reduce la diferencia de presión necesaria para permitir que el pulmón se infle. El cumplimiento del pulmón disminuye y la ventilación disminuye cuando el tejido pulmonar se vuelve enfermo y fibrótico.
Regulación de tamaño alveolar
A medida que los alvéolos aumentan de tamaño, el agente tensoactivo se extiende más por la superficie del líquido. Esto aumenta la tensión superficial reduciendo efectivamente la velocidad de expansión de los alvéolos.
También significa que la velocidad de contracción es más regular, ya que si se reduce el tamaño más rápidamente, la tensión superficial se reducirá más, por lo que otros alvéolos pueden contraerse más fácilmente de lo que pueden.
Prevenir la acumulación de fluidos y mantener las vías respiratorias secas
Las fuerzas de tensión superficial también extraen fluido de los capilares a los espacios alveolares. El tensoactivo reduce la acumulación de fluido y mantiene las vías respiratorias secas al reducir estas fuerzas.
Inmunidad innata
La función inmune del surfactante se atribuye principalmente a dos proteínas: SP-A y SP-D. Estas proteínas se pueden unir a azúcares en la superficie de los patógenos y, por lo tanto, opsonizarlos para su captación por los fagocitos.
La degradación o inactivación del agente tensoactivo puede contribuir a una mayor susceptibilidad a la inflamación e infección pulmonar.
Composición
- ~ 40% de dipalmitoilfosfatidilcolina (DPPC).
- ~ 40% de otros fosfolípidos (PC).
- ~ 10% de proteínas tensioactivas (SP-A, SP-B, SP-C y SP-D).
- ~ 10% de lípidos neutros (colesterol).
- Huellas de otras sustancias.
Lípidos
Dipalmitoilfosfatidilcolina
La dipalmitoilfosfatidilcolina (DPPC) es un fosfolípido con dos cadenas saturadas de 16 carbonos y un grupo fosfato con un grupo amina cuaternaria unida.
la dipalmitoilfosfatidilcolina es la molécula de surfactante más fuerte en la mezcla de surfactante pulmonar.
Esto ocurre principalmente porque la temperatura de transición de fase entre el gel y el cristal líquido de DPPC puro es de 41,5°C, que es más alta que la temperatura del cuerpo humano de 37°C.
Otros fosfolípidos
Las moléculas de fosfatidilcolina forman ~ 85% de los lípidos en el surfactante y tienen cadenas de acilo saturadas. El fosfatidilglicerol (PG), tiene cadenas de ácidos grasos insaturados que fluidizan la monocapa de lípidos en la interfaz.
Los lípidos neutros y el colesterol también están presentes. Los componentes de estos lípidos se difunden desde la sangre a las células alveolares tipo II donde se ensamblan y se empaquetan para la secreción en orgánulos secretores llamados cuerpos lamelares.
Proteínas
Las proteínas constituyen el 10% restante del surfactante. La mitad de este 10% son proteínas plasmáticas, pero el resto está formado por las apolipoproteínas, proteínas tensioactivas SP-A, SP-B, SP-C y SP-D.
Las apolipoproteínas son producidas por la vía secretora en células tipo II. Se someten a mucha modificación postraduccional, terminando en los cuerpos lamelares.
Las proteínas SP-A y SP-D confieren inmunidad innata ya que tienen dominios de reconocimiento de carbohidratos que les permiten recubrir bacterias y virus promoviendo la fagocitosis por los macrófagos.
Las proteínas SP reducen la temperatura crítica de la transición de fase dipalmitoilfosfatidilcolinas a un valor inferior a 37°C, lo que mejora su adsorción y la velocidad de propagación de la interfaz.
Cada proteína SP tiene distintas funciones, que actúan sinérgicamente para mantener una interfaz rica en dipalmitoilfosfatidilcolina durante la expansión y contracción del pulmón.
Entonces las proteínas SP atraen selectivamente más dipalmitoilfosfatidilcolina a la interfaz que otros fosfolípidos o colesterol, cuyas propiedades surfactantes son peores que las dipalmitoilfosfatidilcolinas.
El SP también fija la dipalmitoilfosfatidilcolina en la interfaz para evitar que la dipalmitoilfosfatidilcolina se exprima cuando disminuye el área de la superficie.
Magnitud de la tensión superficial dentro del pulmón
Aunque la tensión superficial puede reducirse en gran medida con el agente tensoactivo pulmonar, este efecto dependerá de la concentración del agente tensoactivo en la interfaz.
La concentración de la interfaz tiene un límite de saturación, que depende de la temperatura y la composición de la mezcla.
Debido a que durante la ventilación existe una variación del área de la superficie del pulmón, la concentración de la interfaz del agente tensoactivo no suele estar en el nivel de saturación.
La superficie aumenta durante la inspiración, lo que abre espacio para que se recluten nuevas moléculas de surfactante en la interfaz.
Mientras tanto, al expirar el área de superficie disminuye, la capa de agente tensoactivo se comprime, acercando las moléculas de agente tensoactivo entre sí y disminuyendo aún más la tensión superficial.
Las moléculas de SP establecen enlaces débiles con las moléculas de surfactante en la interfaz y las mantienen allí durante más tiempo cuando la interfaz está comprimida. Por lo tanto, durante la ventilación, la tensión superficial suele ser menor que en el equilibrio.
Por lo tanto, la tensión superficial varía de acuerdo con el volumen de aire en los pulmones, lo que los protege de la atelectasia a bajos volúmenes y del daño tisular a altos niveles de volumen.
Producción y degradación
La producción de surfactante en humanos comienza en células de tipo II durante la etapa del saco alveolar del desarrollo pulmonar. Los cuerpos lamelares aparecen en el citoplasma alrededor de las 20 semanas de gestación.
Estos cuerpos lamelares se secretan mediante exocitosis en la capa de agua superficial que recubre el espacio alveolar, donde el agente tensoactivo forma una red de mielina tubular.
Se estima que los recién nacidos a término tienen un grupo de almacenamiento alveolar de aproximadamente 100mg/kg de surfactante, mientras que los prematuros tienen un estimado de 4-5 mg/kg al nacer.
Las células Club, también conocidas como células exocrinas bronquiolares, también producen un componente de surfactante pulmonar.
El surfactante alveolar tiene una vida media de 5 a 10 horas una vez secretado. Hasta el 90% del tensoactivo DPPC (dipalmitoil fosfatidilcolina) se recicla desde el espacio alveolar hacia el neumocito tipo II.
Se cree que este proceso ocurre a través de la endocitosis dependiente de clatrina mediada por el receptor estimulante SP-A. El otro 10% es absorbido por los macrófagos alveolares y digerido.
Historia
A finales de la década de 1920, von Neergaard identificó la función del agente tensoactivo pulmonar para aumentar el cumplimiento de los pulmones al reducir la tensión superficial.
También se dio cuenta de la importancia de tener baja tensión superficial en los pulmones de los bebés recién nacidos.
Surfactante pulmonar (medicación)
El surfactante pulmonar se usa como medicamento para tratar y prevenir el síndrome de dificultad respiratoria en bebés recién nacidos. La prevención generalmente se realiza en bebés que nacen con menos de 32 semanas de edad gestacional.
Lo administra el tubo endotraqueal. El inicio de los efectos es rápido. Se pueden necesitar varias dosis.
Los efectos secundarios pueden incluir ritmo cardíaco lento y niveles bajos de oxígeno. Su uso también está relacionado con sangrado intracraneal. El surfactante pulmonar se puede aislar de los pulmones de vacas o cerdos o se puede hacer artificialmente.
El surfactante pulmonar fue descubierto en la década de 1950 y una versión fabricada fue aprobada para uso médico en los Estados Unidos en 1990.
Está en la Lista de Medicamentos Esenciales de la Organización Mundial de la Salud, los medicamentos más efectivos y seguros que se necesitan en un sistema de salud. En el Reino Unido cuesta el NHS 281.64 a 547.40 libras por dosis.
Usos médicos
El surfactante pulmonar se usa para tratar y prevenir el síndrome de dificultad respiratoria en bebés recién nacidos. La prevención generalmente se realiza en bebés que nacen con menos de 32 semanas de edad gestacional. La evidencia tentativa respalda el uso en ahogamiento.
Tipos
Hay varios tipos de surfactantes pulmonares disponibles. Al igual que sus contrapartes naturales, las preparaciones de surfactantes pulmonares consisten en fosfolípidos (principalmente la dipalmitoilfosfatidilcolina) combinados con agentes de expansión tales como SP-B y SP-C.
Tensoactivos pulmonares sintéticos:
- Palmitato de colfoscerilo (Exosurf): una mezcla de dipalmitoilfosfatidilcolina con hexadecanol y tiloxapol agregado como agentes de extensión.
- Pumactant (Compuesto Expansivo de Pulmón Artificial): una mezcla de dipalmitoilfosfatidilcolina y PG.
- KL-4: compuesto de DPPC, palmitoil-oleoil fosfatidilglicerol y ácido palmítico, combinado con un péptido sintético de 21 aminoácidos (sinapultide) que imita las características estructurales de SP-B.
- Venticute: dipalmitoilfosfatidilcolina, PG, ácido palmítico y SP-C recombinante.
Tensoactivos derivados de animales:
- Beractant
- Alveofact: extraído del fluido de lavado pulmonar de la vaca, fabricado por boehringer ingelheim.
- Survanta: extraído del pulmón de vaca picada con dipalmitoilfosfatidilcolina, ácido palmítico y tripalmitina adicionales, fabricado por Abbvie.
- Beraksurf: extraído del pulmón de la vaca picada con dipalmitoilfosfatidilcolina adicional, ácido palmítico y fabricación de tripalmitina por Tekzima.
- Calfactante (Infasurf): extraído del líquido de lavado pulmonar de la pantorrilla.
- Poractant alfa (Curosurf): extraído del material derivado del pulmón de cerdo picado.
El investigador John Clements identificó surfactantes y su papel en la década de 1950. Mary Ellen Avery poco después demostró que los pulmones de los bebés prematuros no podían producir surfactantes.
Exosurf, Curosurf, Infasurf y Survanta fueron los tensoactivos iníciales aprobados por la Administración de Alimentos y Drogas para su uso en EE. UU. En 2012, la Administración de Alimentos y Drogas de EE. UU. Aprobó un surfactante sintético adicional, lucinactant (Surfaxin).
Enfermedades
La deficiencia de surfactante pulmonar es la principal causa del síndrome de dificultad respiratoria en los bebés prematuros. Se han descrito cuatro proteínas asociadas al agente tensoactivo, las proteínas surfactantes A, B, C y D, y dos se han asociado con enfermedades difusas del parénquima pulmonar.
La proteína surfactante C (SP-C) es una proteína altamente hidrofóbica que mejora las propiedades de disminución de la tensión superficial del surfactante pulmonar.
Los casos familiares de dificultad respiratoria neonatal se han asociado con la deficiencia de proteína B surfactante, pero la dificultad respiratoria de los recién nacidos no se considera una forma de enfermedades pulmonares parenquimatosas difusas/enfermedad pulmonar intersticial.
Las variantes genéticas de SP-A se han asociado con un mayor riesgo de fibrosis pulmonar idiopática.
Deficiencia congénita de surfactante
La disfunción del metabolismo del surfactante es una condición donde el surfactante pulmonar es insuficiente para la respiración adecuada.
La mayoría de las mutaciones causantes de enfermedades en SFTPB dan como resultado una falta completa de proteína SP-B madura 265120. La enfermedad pulmonar se hereda de manera autosómica recesiva, requiriendo mutaciones en ambos alelos.
El surfactante producido por bebés con deficiencia de SP-B es anormal en su composición y no funciona normalmente al disminuir la tensión superficial.
Los casos familiares de disfunción SP-C 610913 se heredan en un patrón autosómico dominante, aunque el inicio y la gravedad de la enfermedad pulmonar son muy variables, incluso dentro de la misma familia.
Las mutaciones en ABCA3 parecen ser la causa más común de disfunción de surfactante genético en humanos. Las mutaciones dan como resultado una pérdida o una función reducida de la proteína ABCA3, y se heredan de forma autosómica recesiva 610921.
Proteinosis alveolar pulmonar
La proteinosis alveolar pulmonar (PAP) es un grupo de trastornos pulmonares raros caracterizados por la acumulación anormal de compuestos de lipoproteína derivados de surfactante dentro de los alvéolos del pulmón.
Las sustancias acumuladas interfieren con el intercambio normal de gases y la expansión de los pulmones, lo que en última instancia provoca dificultad para respirar y una predisposición a desarrollar infecciones pulmonares.
Las causas de la proteinosis alveolar pulmonar se pueden agrupar en causas primarias y secundarias, aunque la etiología más común es una afección autoinmune primaria.