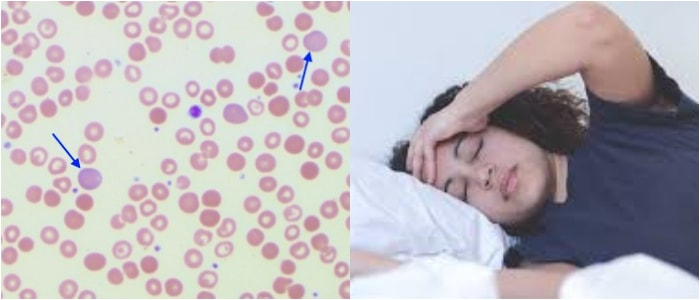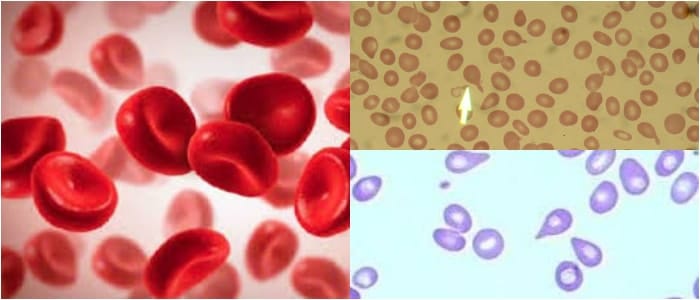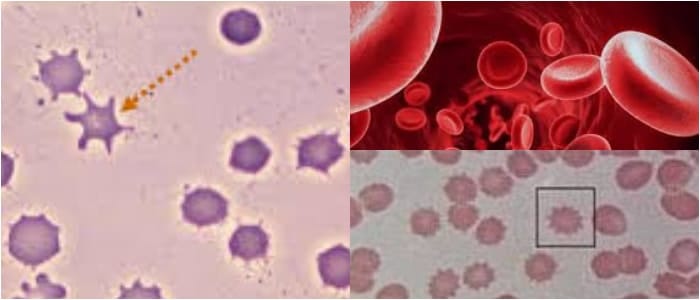
Son hematíes cuya forma se parece a la de las hojas de acanto.
Esta planta mediterránea se caracteriza por el tamaño importante de sus hojas. Dotados de múltiples espículas y proyecciones en superficie, estos glóbulos rojos constituyen una anomalía que se asocia, generalmente, a unos lípidos que tienen un metabolismo anormal.
Los acantocitos se forman cuando los ácidos grasos libres se fijan en cantidades importantes sobre la membrana eritrocitaria de los hematíes.
Los acantocitos se encuentran en la sangre de individuos con ChAc en una proporción muy variable, generalmente del 5% al 50% de la población de glóbulos rojos.
En algunos casos, la acantocitosis puede estar ausente o puede aparecer solo tarde en el curso de la enfermedad.
La proporción de acantocitos no se correlaciona con la gravedad de la enfermedad.
Detección de los acantocitos
La microscopía electrónica de barrido de eritrocitos fijados con glutaraldehído es probablemente el método más confiable para detectar acantocitos, pero no está disponible de manera rutinaria.
Se ha propuesto un estándar general para la determinación de acantocitosis.
La sangre se diluye 1: 1 con solución salina al 0,9% y 10 U / ml de heparina, y se examina usando microscopía de contraste de fase después de 30 minutos de incubación en un agitador.
En muestras normales, menos del 6,3% de las células están espiculadas [ Storch et al 2005 ]. (Los frotis de sangre seca a menudo son inadecuados).
Acantocitosis
La acantocitosis ocurre debido a anormalidades ultraestructurales del esqueleto membranoso de los eritrocitos que resultan en una reducción de la fluidez de la membrana.
Al menos tres afecciones neurológicas hereditarias están asociadas con él, aunque todavía se desconoce la patogénesis de las características neurológicas.
En la abetalipoproteinemia, una afección autosómica recesiva, la deficiencia de vitamina E produce un síndrome espinocerebeloso progresivo asociado con neuropatía periférica y retinitis pigmentosa.
La neuroacantocitosis también es probablemente una afección autosómica recesiva y se caracteriza por corea, discinesia orofaciolingual, disartria, arreflexia, convulsiones y demencia.
Detección de corea-acantocitosis (ChAc)
El análisis de transferencia Western reveló ausencia o reducción marcada de la coreoína, la proteína codificada por VPS13A , en eritrocitos de individuos con ChAc.
En contraste, se observaron niveles normales de coreoína en muestras de individuos con síndrome de McLeod y enfermedad de Huntington , lo que sugiere que la pérdida de coreaina completa es un diagnóstico de ChAc [ Dobson-Stone et al 2004 ].
Es de destacar que los niveles normales de coreoina son teóricamente posibles para algunos alelos variantes patogénicos de VPS13A (por ejemplo, algunos erroressustituciones); por lo tanto, la presencia de niveles normales de coreoína no excluye el diagnóstico de ChAc.
Características clínicas
La corea-acantocitosis (ChAc) se caracteriza por un trastorno del movimiento progresivo, cambios cognitivos y de comportamiento, una miopatía que puede ser subclínica e hiperCquemia crónica en suero.
Aunque el trastorno recibe el nombre de acantocitosis de los glóbulos rojos, esta característica es variable. El trastorno del movimiento es principalmente corea de las extremidades, pero algunas personas presentan parkinsonismo.
La distonía es común y afecta la región oral y especialmente la lengua, causando disartria y disfagia grave con la consiguiente pérdida de peso. Las características habituales de morderse la lengua y los labios son características, así como la distonía por protrusión de la lengua.
Los cambios cognitivos y conductuales progresivos se parecen a los de un síndrome del lóbulo frontal. Se observan convulsiones en casi la mitad de los afectadosindividuos y puede ser la manifestación inicial. La miopatía produce un desgaste progresivo del músculo distal y debilidad.
La edad media de aparición en ChAc es de aproximadamente 30 años, aunque ChAc puede desarrollarse tan pronto como la primera década o tan tarde como la séptima década.
Lleva a cabo un curso progresivo crónico y puede conducir a una discapacidad importante en unos pocos años. La esperanza de vida se reduce, con una edad de muerte que oscila entre 28 y 61 años.
Diagnóstico / prueba
El diagnóstico de ChAc se basa principalmente en hallazgos clínicos, la presencia de hallazgos característicos de MRI y evidencia de enfermedad muscular. La CT y la MRI revelan atrofia de los núcleos caudados con dilatación de los cuernos anteriores de los ventrículos laterales.
La resonancia magnética comúnmente muestra un aumento de la señal ponderada en T 2 en el caudado y el putamen. Los acantocitos están presentes en 5% -50% de la población de glóbulos rojos.
En algunos casos, la acantocitosis puede estar ausente o puede aparecer solo tarde en el curso de la enfermedad. Se observa un aumento de la concentración sérica de creatina quinasa muscular (CK) en la mayoría de los individuos afectados.
La biopsia muscular revela núcleos centrales y fibras atróficas. VPS13A, que codifica la coreina, es el único gen en el que actualmente se sabe que la mutación causa ChAc.
Administración
Tratamiento de las manifestaciones:
El tratamiento es puramente sintomático y puede incluir:
- Toxina botulínica para disminuir la distonía orofacio-lingual.
- Asistencia de alimentación.
- Terapia del lenguaje.
- Dispositivos de protección mecánica.
- Férulas para la caída del pie.
- Fenitoína, clobazam, valproato y levetiracetam para el manejo de las convulsiones.
- Medicamentos antidepresivos o antipsicóticos.
- Antagonistas / agotadores de dopamina tales como neurolépticos atípicos o tetrabenazina.
La estimulación cerebral profunda puede ser útil en casos seleccionados.
- Vigilancia: monitoreo del estado nutricional y adaptación de la dieta para asegurar una ingesta calórica adecuada y evitar la aspiración; EEG cada tercer año.
- Agentes / circunstancias a evitar: circunstancias que provocan convulsiones y anticonvulsivos que pueden empeorar los movimientos involuntarios.
Asesoramiento genético
La ChAc se hereda de manera autosómica recesiva . En la concepción, cada hermano de un individuo afectado tiene una probabilidad del 25% de verse afectado, una probabilidad del 50% de ser un portador asintomático y una probabilidad del 25% de no verse afectado y no ser portador.
La prueba de portador para parientes en riesgo es posible si se conocen las variantes patogénicas en la familia. Las pruebas prenatales son posibles para familias en las que se conocen las variantes patogénicas.