
Es un raro trastorno genético multisistémico.
Es caracterizado por la acumulación de un aminoácido llamado cistina en diferentes tejidos y órganos del cuerpo, incluidos los riñones, los ojos, los músculos, el hígado, el páncreas y el cerebro.
En general, la cistinosis se divide en tres formas diferentes conocidas como cistinosis nefropática, cistinosis intermedia y cistinosis no nefropática (u ocular).
La edad de inicio, los síntomas y la gravedad de la cistinosis pueden variar mucho de una persona a otra. La cistinosis nefropática se presenta en la infancia y es la forma más común y grave.
La detección temprana y el tratamiento oportuno son críticos para reducir el desarrollo y la progresión de los síntomas asociados con la cistinosis. Los riñones y los ojos son los dos órganos más afectados.
Las personas con cistinosis nefropática o intermedia en última instancia, requieren un trasplante de riñón.
La cistinosis no nefropática solo afecta las córneas de los ojos. La cistinosis es causada por mutaciones del Gen CTNS y se hereda como una enfermedad autosómica recesiva.
La cistinosis fue descrita por primera vez en la literatura médica en 1903 por Abderhalden. La cistinosis se clasifica como un trastorno de almacenamiento lisosomal.
Los lisosomas son compartimentos unidos a la membrana dentro de las células que descomponen ciertos nutrientes como las grasas, las proteínas y los carbohidratos.
Los lisosomas son la unidad digestiva primaria dentro de las células. Algunas enzimas dentro de los lisosomas descomponen (metabolizan) estos nutrientes, mientras que otras enzimas transportan los productos metabólicos sobrantes (como la cistina) fuera del lisosoma.
En el caso de la cistinosis, la falta de un transportador específico de este tipo hace que la cistina se acumule en los lisosomas de las células de todo el cuerpo. La cistina forma cristales (cristaliza) en muchos tipos de células y daña lentamente los órganos afectados.
Signos y síntomas
En un momento, la cistinosis nefropática fue fatal a una edad muy temprana.
Sin embargo, el desarrollo de un medicamento conocido como cisteamina (que reduce los niveles de cistina en el cuerpo) y las mejoras en los trasplantes de riñón han transformado la cistinosis de un trastorno renal fatal a un trastorno multisistémico crónico con una expectativa de vida que llega hasta la edad adulta e incluso más allá.
Los síntomas específicos y la gravedad de la cistinosis varían mucho de una persona a otra en función de varios factores, incluida la edad de aparición y si el trastorno se diagnostica y trata de inmediato.
La progresión del trastorno puede ser retardada por el diagnóstico y tratamiento tempranos. Eventualmente, la cistinosis puede afectar todos los tejidos del cuerpo. La edad de inicio para diferentes síntomas varía enormemente.
Es importante tener en cuenta que las personas afectadas pueden no tener todos los síntomas que se analizan a continuación.
Las personas afectadas y los padres de los niños afectados deben hablar con su médico y equipo médico sobre su caso específico, los síntomas asociados y el pronóstico general.
Cistinosis nefropática
La cistinosis nefropática o infantil es la forma más frecuente y más grave de cistinosis. Los síntomas de la cistinosis nefropática generalmente se manifiestan en la segunda mitad del primer año de vida.
Los síntomas específicos pueden ser leves o graves según cada caso individual y la edad en que se inicia el tratamiento.
El fracaso del crecimiento y el síndrome de Fanconi renal suelen ser las primeras complicaciones notables del trastorno. Aunque los bebés parecen normales al nacer, a la edad de un año a menudo caen en el tercer percentil para la estatura y el peso.
Además, los bebés afectados pueden tener episodios de vómitos, falta de apetito y dificultades con la alimentación que contribuyen (junto con la disfunción renal) a la deficiencia nutricional y la falta de aumento de peso y crecimiento a la tasa esperada (falta de crecimiento).
En promedio, el crecimiento en niños no tratados con cistinosis ocurre a un 60 por ciento de la tasa esperada.
Los bebés con cistinosis nefropática desarrollan el síndrome renal de Fanconi, un trastorno raro caracterizado por disfunción renal. Los riñones son dos órganos con forma de frijol ubicados justo debajo de la caja torácica.
Los riñones tienen varias funciones, como filtrar y excretar los productos de desecho de la sangre y el cuerpo, crear ciertas hormonas y ayudar a mantener el equilibrio de ciertos químicos en el cuerpo como el potasio, sodio, cloruro, calcio, magnesio y otros minerales y electrolitos.
En la cistinosis nefropática, los túbulos renales no pueden reabsorber una variedad de sustancias necesarias, incluidos los compuestos mencionados anteriormente, así como los aminoácidos, fosfato, calcio, glucosa, carnitina, ciertas proteínas y electrolitos.
En consecuencia, los individuos afectados tienen niveles anormalmente bajos de muchas de estas sustancias en el cuerpo.
Los síntomas del síndrome de Fanconi renal generalmente se manifiestan entre los 6 y 12 meses de edad y pueden incluir sed excesiva (polidipsia), producción y paso de orina excesivos (poliuria), desequilibrios electrolíticos, vómitos y deshidratación con o sin fiebre.
La deshidratación puede ser grave en algunas personas afectadas.
El síndrome renal de Fanconi también puede causar raquitismo hipofosfatémico. En este trastorno, debido a que los riñones no pueden reabsorber el fósforo de la orina, el cuerpo filtra el fosfato de los huesos para mantener los niveles de fósforo en la sangre; esto resulta en un ablandamiento y debilitamiento progresivos del hueso (raquitismo).
El raquitismo puede causar deformidad ósea y retrasar el caminar, porque es doloroso. Los niños afectados pueden caminar con cautela. La disfunción renal también puede causar que se pierdan cantidades excesivas de calcio en el cuerpo a través de la orina (hipocalcemia hipercalciúrica).
Los niveles bajos de calcio pueden causar espasmos musculares intermitentes (tetania) y, rara vez en la cistinosis, convulsiones.
Si no se trata, la función de filtración del riñón continuará deteriorándose, y eventualmente progresará a una insuficiencia renal a los 10 años de edad.
El tratamiento con medicamentos que reducen los niveles de cistina puede retardar o detener la progresión de la enfermedad renal y retrasar la necesidad de un trasplante de riñón en los años de la adolescencia, hasta los 20 años o más.
Cualquier daño renal existente que ocurra antes del diagnóstico (y, por lo tanto, antes del tratamiento) es irreversible.
Síntomas extrarrenales
Los niños con cistinosis nefropática también pueden desarrollar síntomas no relacionados con los riñones (síntomas extrarrenales). Nuevamente, estos hallazgos son muy variables y un niño afectado no desarrollará todos los síntomas que se analizan a continuación.
Los síntomas extrarrenales específicos varían mucho según la edad en que se inicie el tratamiento y los órganos específicos que se involucren; esos órganos pueden incluir:
- Los ojos.
- La médula ósea.
- El hígado.
- El páncreas.
- El bazo.
- El intestino.
- El cerebro.
- La tiroides.
- Los músculos.
- Los testículos.
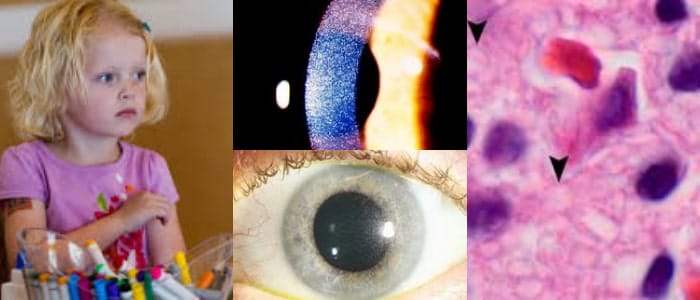
A cualquier edad, los niños pueden desarrollar una sensibilidad anormal a la luz (fotofobia) e irritación debido a la formación de cristales de cistina en la córnea. La severidad de la fotofobia puede variar. En algunos individuos no tratados, pueden desarrollarse dolor y erosiones corneales recurrentes.
Alrededor de los 10 años, los niños afectados también pueden desarrollar una deficiencia de la producción de hormona tiroidea (hipotiroidismo) debido a la acumulación de cristales de cistina en la tiroides. La tiroides es una glándula con forma de mariposa ubicada en la base del cuello.
La tiroides secreta hormonas en el torrente sanguíneo que influyen en ciertas actividades del cuerpo, como el crecimiento, la maduración y la tasa de metabolismo. Los síntomas del hipotiroidismo son muy variables, pero pueden incluir fatiga, sensación de frío, sequedad de la piel, estreñimiento y depresión.
Como grupo, los niños con cistinosis nefropática no producen cantidades normales de lágrimas, sudor o saliva. La producción de lágrimas puede disminuir y los ojos se secan.
Una capacidad disminuida para sudar puede potencialmente causar agotamiento total o colapso debido al calor (postración por calor).
La pubertad puede demorarse uno o dos años. Los varones no tratados experimentan hipogonadismo, en el que los testículos producen cantidades reducidas de testosterona.
La testosterona desempeña un papel clave en el crecimiento y el desarrollo de las características sexuales secundarias masculinas durante la pubertad.
La inteligencia suele ser normal, aunque muchos niños experimentan discapacidades de aprendizaje.
Algunos niños pueden tener problemas con el procesamiento de la información visual, la memoria visual a corto plazo, las dificultades para identificar objetos comunes mediante el tacto (reconocimiento táctil) y la incapacidad de reconocer visualmente la relación espacial entre los objetos (habilidades visoespaciales deficientes).
Un ejemplo de habilidades visuoespaciales es la percepción de la distancia y la profundidad. También se han reportado problemas con la velocidad del motor y la atención sostenida.
Algunos niños afectados presentan problemas de comportamiento y psicosociales, que son comunes en niños que padecen enfermedades crónicas.
Los niveles de CI, mientras que en el rango normal, pueden ser más bajos de lo que se esperaría en base a los niveles de CI de padres y hermanos.
Los niños con cistinosis nefropática pueden presentar rasgos faciales levemente alterados (dismorfología craneofacial). También puede ocurrir un desarrollo dental tardío y una erupción retardada de los dientes permanentes.
Algunas personas afectadas pueden desarrollar un aumento de la presión del líquido cefalorraquídeo dentro del cerebro (hipertensión intracraneal), que puede causar dolores de cabeza e hinchazón del disco óptico (papiledema).
Anomalías de inicio tardío
El aumento de la longevidad de las personas con cistinosis nefropática ha revelado que pueden aparecer complicaciones adicionales que afectan a otros órganos además de los riñones durante la vida.
Estas complicaciones se desarrollan debido a la acumulación crónica de cristales de cistina en individuos que no han sido tratados adecuadamente con cisteamina, aunque se han sometido a un trasplante de riñón. Estas complicaciones adicionales generalmente se desarrollan entre los 20 y 40 años de edad.
La acumulación de cistina en el tejido muscular puede causar una enfermedad muscular (miopatía) que conduce a una debilidad progresiva y al desgaste de los músculos afectados.
El deterioro de los músculos de la garganta puede llevar a dificultades para tragar y alimentar. La afectación de los músculos del tórax puede provocar insuficiencia pulmonar.
Se puede desarrollar una amplia variedad de síntomas gastrointestinales, como:
- Agrandamiento del hígado (hepatomegalia), presión arterial alta de la vena principal del hígado (hipertensión portal).
- Agrandamiento del bazo (esplenomegalia).
- Reflujo gastroesofágico.
- Úlceras, inflamación del esófago (esofagitis).
- Disfunción de los músculos del tracto gastrointestinal (dismotilidad).
Los síntomas adicionales inusuales incluyen:
- Enfermedad inflamatoria intestinal.
- Desgarro del intestino que hace que los contenidos del intestino fluyan hacia la cavidad abdominal (perforación intestinal).
- Inflamación del peritoneo (peritonitis), que es la membrana que recubre la pared abdominal y los órganos.
La presión arterial alta (hipertensión), la aterosclerosis de las arterias coronarias y las anomalías de la coagulación de la sangre son complicaciones de la enfermedad renal asociada con la cistinosis.
Los hallazgos adicionales incluyen la enfermedad ósea metabólica y la incapacidad de digerir correctamente los alimentos debido a la falta de enzimas digestivas producidas normalmente por el páncreas (insuficiencia pancreática exocrina).
Los adultos con cistinosis también pueden desarrollar anomalías oculares, como espasmos de los párpados (blefarospasmo), queratopatía en banda y retinopatía pigmentaria.
La queratopatía en banda se refiere a la acumulación de depósitos de calcio en una banda a través de la superficie central de la córnea, que puede causar dolor y disminución de la claridad de la visión (agudeza visual).
La retinopatía pigmentaria se caracteriza por una degeneración progresiva de la retina, la capa delgada de las células nerviosas que recubren la superficie interna de la parte posterior de los ojos.
La retina percibe la luz y la convierte en señales nerviosas, que luego se transmiten al cerebro a través del nervio óptico.
Aunque es poco común, la disfunción cerebral ocurre en algunos adultos mayores con cistinosis. La razón exacta por la que esto ocurre es desconocida.
Los síntomas específicos variarán, pero algunos individuos afectados pueden experimentar una disminución en las capacidades motoras y mentales. En casos muy raros, la disfunción neurológica puede progresar a la demencia.
Las personas con cistinosis nefropática parecen tener una mayor tasa de diabetes que la población general debido a la destrucción del páncreas por la acumulación de cistina.
Cistinosis intermedia
También conocida como cistinosis juvenil nefropática o cistinosis adolescente, esta forma de cistinosis se caracteriza por todos los signos y síntomas de la cistinosis nefropática descritos anteriormente.
Sin embargo, la aparición de estos síntomas no se produce hasta más tarde, tal vez a los 8-20 años de edad.
En general, los síntomas son menos severos que en la forma nefropática infantil clásica y tienen una progresión más lenta. Si no se trata, la insuficiencia renal terminal en la cistinosis intermedia generalmente se desarrolla en algún momento entre los 15 y los 25 años de edad.
Existe un espectro de gravedad de la enfermedad en la cistinosis, con superposición de las formas infantiles e intermedias.
Cistinosis no nefropática
También conocida como cistinosis ocular o “benigna”, esta forma generalmente afecta a los adultos durante la mediana edad; Una vez fue llamada cistinosis adulta. La enfermedad renal no se produce en estos individuos.
El trastorno parece afectar solo a los ojos. Los individuos no tratados con cistinosis no nefropática eventualmente desarrollan fotofobia debido a la acumulación de cristales de cistina en los ojos.
Causas
Todos los tipos de cistinosis son causados por mutaciones del CTNS.gene. La enfermedad se hereda de forma autosómica recesiva.
Los trastornos genéticos recesivos ocurren cuando un individuo hereda una mutación en el mismo gen de cada padre. Si un individuo recibe un gen normal y un gen para la enfermedad, la persona será portadora de la enfermedad, pero por lo general no mostrará síntomas.
Los portadores de la cistinosis (p. Ej., Los padres de las personas afectadas) nunca tienen síntomas de la enfermedad. El riesgo de que dos padres portadores transmitan el gen defectuoso y, por lo tanto, tenga un hijo afectado es del 25 por ciento con cada embarazo.
El riesgo de tener un hijo que es portador como los padres es del 50 por ciento con cada embarazo. La posibilidad de que un niño reciba genes normales de ambos padres y sea genéticamente normal para ese rasgo en particular es del 25 por ciento. El riesgo es el mismo para hombres y mujeres.
Los investigadores han determinado que el gen CTNS se encuentra en el brazo corto (p) del cromosoma 17 (17p13). Los cromosomas, que están presentes en el núcleo de las células humanas, llevan la información genética de cada individuo. Las células humanas normalmente tienen 46 cromosomas.
Los pares de cromosomas humanos están numerados del 1 al 22 y los cromosomas sexuales se designan como X e Y. Los hombres tienen un cromosoma X y uno Y y las mujeres tienen dos cromosomas X.
Cada cromosoma tiene un brazo corto designado como «p» y un brazo largo designado como «q».
Los cromosomas se subdividen en muchas bandas que están numeradas. Por ejemplo, «cromosoma 17p13» se refiere a la banda 13 en el brazo corto del cromosoma 17. Las bandas numeradas especifican la ubicación aproximada de los miles de genes que están presentes en cada cromosoma.
El gen CTNS contiene instrucciones para producir (codificar) una proteína llamada cistinosina que se requiere para transportar el aminoácido cistina fuera de los lisosomas y al resto de una célula.
Los lisosomas descomponen (degradan) ciertas proteínas en aminoácidos de sus componentes, como la cistina.
La cistina es transportada fuera del lisosoma por la cistinosina. Los niveles deficientes de cistinosina funcional dan como resultado la acumulación (almacenamiento) de cistina en los lisosomas de diversos tejidos y órganos del cuerpo.
La cistina acumulada forma cristales, que eventualmente dañan los órganos afectados.
Poblaciones afectadas
La cistinosis afecta a hombres y mujeres en igual número. Se estima que el trastorno ocurre en 1 de cada 100,000-200,000 personas en la población general. La cistinosis ha sido reportada en todo el mundo, en todos los grupos étnicos.
La cistinosis es la causa más común del síndrome de Fanconi renal en niños y representa aproximadamente el 5 por ciento de todos los casos infantiles de insuficiencia renal.
Trastornos relacionados
Los síntomas de los siguientes trastornos pueden ser similares a los de la cistinosis. Las comparaciones pueden ser útiles para un diagnóstico diferencial.
Muchos trastornos diferentes pueden causar el síndrome de Fanconi renal en niños como el síndrome de Lowe, la enfermedad de Wilson, la tirosinemia tipo I, la galactosemia y las enfermedades por almacenamiento de glucógeno.
Los trastornos adicionales que pueden tener síntomas similares a los encontrados en la cistinosis incluyen el síndrome de Bartter y la diabetes insípida.
Hay varios tipos de trastornos metabólicos en los cuales la acumulación secundaria de ciertas sustancias como las grasas y los carbohidratos se produce en el cuerpo.
Estos trastornos incluyen:
- Galactosemia.
- Sialidosis.
- Enfermedad de Gaucher.
- Galactosialidosis.
- Enfermedad de Wolman.
- Enfermedad de almacenamiento de éster de colesterilo.
- Mucopolisacaridosis.
- Otros trastornos de almacenamiento lisosomal.
Diagnóstico
Un diagnóstico de cistinosis se basa en la identificación de los síntomas característicos (p. Ej., Síntomas del síndrome de Fanconi renal), una historia detallada del paciente, una evaluación clínica completa y una variedad de pruebas especializadas.
Un diagnóstico rápido de la cistinosis es fundamental para maximizar los beneficios preventivos y terapéuticos de los medicamentos que agotan la cistina.
Pruebas clínicas y exámenes
El diagnóstico de la cistinosis se puede confirmar midiendo los niveles de cistina en ciertos glóbulos blancos («leucocitos polimorfonucleares»).
El examen urinario puede revelar una pérdida excesiva de nutrientes, incluidos minerales, electrolitos, aminoácidos, carnitina y agua, lo cual es indicativo del síndrome renal de Fanconi.
Un médico puede usar un microscopio especial llamado lámpara de hendidura para ver los ojos con un gran aumento, que puede revelar cristales de cistina en la córnea. Esto es diagnóstico si lo realiza un oftalmólogo con experiencia.
Un diagnóstico de cistinosis se puede confirmar mediante pruebas genéticas moleculares, que pueden identificar la mutación característica del gen CTNS que causa el trastorno. Las pruebas genéticas moleculares están disponibles a través de un laboratorio comercial.
El diagnóstico prenatal está disponible para familias con un riesgo conocido de tener un bebé con cistinosis. Los niveles de cistina pueden medirse en células obtenidas del fluido que rodea al feto en desarrollo (líquido amniótico).
Una prueba conocida como muestreo de vellosidades coriónicas también se puede usar para obtener un diagnóstico prenatal de cistinosis. Las vellosidades coriónicas son estructuras delgadas similares a pelos que se encuentran en la placenta.
Estas células se pueden examinar para detectar niveles elevados de cistina.
Tratamiento
El tratamiento de la cistinosis se dirige hacia los síntomas específicos que son evidentes en cada individuo. El tratamiento puede requerir los esfuerzos coordinados de un equipo de especialistas.
Los pediatras, los especialistas en riñones (nefrólogos), los especialistas de la vista (oftalmólogos), los especialistas en trastornos digestivos (gastroenterólogos), los psicólogos y otros profesionales de la salud pueden necesitar un plan sistemático y completo para el tratamiento del niño afectado.
La cistinosis nefropática e intermedia fue una vez trastornos progresivamente fatales, con una vida útil para la forma infantil de menos de 10 años. Sin embargo, el desarrollo de terapias que agotan la cistina junto con las mejoras en el trasplante de riñón han extendido la vida útil hasta la edad adulta.
Terapia de agotamiento de la cistina
En 1994, la Administración de Alimentos y Medicamentos (FDA) aprobó el bitartrato de cisteamina (Cystagon®) para el tratamiento de personas con cistinosis. En 2013, la FDA aprobó Procysbi®, una forma de liberación prolongada de cisteamina.
La cisteamina es un agente reductor de la cistina que puede disminuir considerablemente los niveles de cistina dentro de las células. La terapia con cisteamina retarda el desarrollo y la progresión del daño renal y mejora el crecimiento en los niños.
La cisteamina puede retrasar significativamente la necesidad de un trasplante de riñón. Algunas personas que se sometieron a un tratamiento temprano y diligente con cisteamina pudieron retrasar un trasplante de riñón hasta los 20 años o más.
La terapia con cisteamina debe iniciarse inmediatamente después del diagnóstico para prevenir o retardar el daño renal. La cisteamina debe continuarse durante toda la vida porque los estudios han demostrado que el tratamiento prolongado con cisteamina y puede prevenir muchas de las complicaciones no renales y tardías de la cistinosis.
La cisteamina se toma por vía oral, pero la cisteamina oral no llega a la córnea con eficacia y, por lo tanto, no logra eliminar los cristales de cistina de la córnea.
El Cystaran® (Sigma-Tau Pharmaceuticals) fue aprobado por la FDA en 2012 como un tratamiento para la acumulación de cristales de cistina en la córnea asociada con la cistinosis.
Esta solución de gotas oculares que contiene cisteamina es el único tratamiento aprobado por la FDA para las complicaciones oculares de este trastorno.
La terapia con cisteamina puede causar náuseas, vómitos y molestias gastrointestinales. La cisteamina también causa la secreción excesiva de ácidos del estómago.
Es posible que algunas personas que toman cisteamina deban tomar inhibidores de la bomba de protones, como el omeprazol, para reducir la producción de ácido estomacal, lo que ayuda a mejorar los síntomas gastrointestinales.
La cisteamina huele y sabe mal, lo que a veces conduce a problemas de cumplimiento con las personas afectadas.
Terapia sintomática
El síndrome de Fanconi renal se trata con una ingesta alta de líquidos y electrolitos para prevenir la reducción excesiva del agua corporal (deshidratación).
Se puede administrar bicarbonato de sodio, citrato de sodio, magnesio y potasio para ayudar a mantener el equilibrio normal de electrolitos. Los inhibidores de la acetilcolinesterasa (ACE) a veces se usan con la esperanza de disminuir la progresión de la enfermedad renal.
La indometacina es un medicamento antiinflamatorio que a veces se usa para reducir las pérdidas urinarias de agua y electrolitos; A veces también ayuda a mejorar la tasa de crecimiento. Si los individuos afectados toman indometacina, deben ser vigilados estrechamente con respecto a su función renal.
Los fosfatos y la vitamina D a menudo se administran para corregir la reabsorción dañada de fosfato en la sangre y prevenir el raquitismo. La carnitina se puede prescribir para algunas personas antes del trasplante para mejorar la fuerza muscular.
Una nutrición adecuada es de vital importancia para garantizar que los bebés y niños afectados maximicen su potencial de crecimiento. La terapia con hormona de crecimiento ha mejorado significativamente el crecimiento en muchos pacientes.
La L-tiroxina se usa para tratar el hipotiroidismo, la insulina para tratar la diabetes dependiente de la insulina y la testosterona para tratar a los hombres con función testicular poco activa (hipogonadismo) para que se desarrollen características sexuales secundarias.
La infertilidad no responderá al tratamiento con testosterona.
Los síntomas oculares de la cistinosis se pueden tratar evitando la luz brillante, las gafas de sol y la lubricación. En casos extremadamente raros, puede ser necesario un trasplante de córnea.
Por lo general, esto solo se requiere para personas con queratopatías de banda grande o aquellas que sufren dolor debido a la erosión corneal repetida.
Algunos bebés y niños con cistinosis (por ejemplo, aquellos con disfagia, mala nutrición y mayor riesgo de aspiración) pueden requerir la implantación de un tubo de gastronomía.
Con este procedimiento, se coloca un tubo delgado en el estómago a través de una pequeña incisión en el abdomen, lo que permite la ingesta directa de alimentos y / o medicamentos.
La terapia del habla y el lenguaje puede ser beneficiosa en algunos casos. La asesoría genética puede ser beneficiosa para las personas afectadas y sus familias.
Trasplante renal
A pesar del tratamiento temprano y rápido, los individuos con cistinosis infantil e intermedia eventualmente desarrollan enfermedad renal en etapa terminal (ESRD), que requiere un trasplante de riñón. Inicialmente, una persona afectada puede someterse a diálisis.
La diálisis es un procedimiento en el que se utiliza una máquina para realizar algunas de las funciones de los riñones: filtrar los productos de desecho del torrente sanguíneo y ayudar a mantener niveles adecuados de sustancias químicas esenciales, como el potasio.
La ESRD no es reversible, por lo que las personas eventualmente requerirán un trasplante de riñón. La tasa de progresión de la disfunción renal a la ESRD puede variar mucho de un individuo a otro.
Las personas con cistinosis generalmente responden muy bien a un trasplante de riñón, que puede curar el síndrome de Fanconi renal porque la cistina no se acumula en el riñón donado.


